#2 Chromatin remodeling correlates with transcription changes
Mireia Ramos-Rodríguez
Details
Original publication:
Colli, M.L., Ramos-Rodríguez, M., Nakayasu, E.S. et al. An integrated multi-omics approach identifies the landscape of interferon-α-mediated responses of human pancreatic beta cells. Nat Commun 11, 2584 (2020). https://doi.org/10.1038/s41467-020-16327-0
Contents: Analyses and figures contained in this document correspond to the following figures/sections of the original publication:
- Results: “Interferon-\(\alpha\) induces early changes in chromatin accessibility”.
- Figure 1: “Exposure of EndoC-\(\beta\)H1 cells to interferon-\(\alpha\) promotes changes in chromatin accessibility, which are correlated with gene transcription and translation”. Panel c and d.
- Supplementary Figure 3: “Gained open chromatin regions are mainly localized distally to gene transcription starting sites (TSSs), evolutionary conserved and enriched in transcription factors (TFs) binding motifs”. Panel e.
Analysis of RNA-seq data
Differential analysis
Using Rsubread and DESeq2, we can easily generate the list of open regions in the samples, including the normalized counts, adjusted p-values and log2 Fold Changes. The steps that we have to follow are:
- Obtain counts per gene using RSubread::featureCounts. (! Careful with the arguments, check that they are correct). We annotate to ENSEMBL GRCh37 GTF
Homo_sapiens.GRCh37.87.gtf - Create
DESeqDataSetobject including annotation data. - Perform differential analysis, obtain results and counts and save into a
data.frame.
Note: This differential analysis of RNA-seq data was only used to correlate gene expression with open chromatin. For the methods used throughout the paper to detect differential gene expression, please see Supplementary Methods section RNA sequencing processing and analysis, in Colli et al, 2020.
###############################################################################
## 1) Obtain counts for each gene ---------------------------------------------
anno <- "/home/labs/lplab/mramos/data/Homo_sapiens.GRCh37.87.gtf"
files <- list.files("../../data/RNA-seq/BAMs", full.names=TRUE,
pattern="*.bam$")
bam_files_2h <- files[grep("_2h_", files)]
bam_files_24h <- files[grep("_24h_", files)]
bam_files_8h <- files[grep("_8h_", files)]
counts_2h <- Rsubread::featureCounts(bam_files_2h,
annot.ext=anno,
isGTFAnnotationFile=TRUE,
isPairedEnd=TRUE,
nthreads=6)
counts_8h <- Rsubread::featureCounts(bam_files_8h,
annot.ext=anno,
isGTFAnnotationFile=TRUE,
isPairedEnd=TRUE,
nthreads=6)
counts_24h <- Rsubread::featureCounts(bam_files_24h,
annot.ext=anno,
isGTFAnnotationFile=TRUE,
isPairedEnd=TRUE,
nthreads=6)
###############################################################################
## 2) Perform differential analysis -------------------------------------------
## 2 hours -----------------------------------
## Create DDS object and perform analysis
cts <- counts_2h$counts
colnames(cts) <- names_2h
coldata <- data.frame("treatment"=gsub("_[0-9]$", "", names_2h))
dds_2h <- DESeqDataSetFromMatrix(countData = cts,
colData = coldata,
design= ~ treatment)
## Run analysis in parallel
library("BiocParallel")
register(MulticoreParam(6))
dds_2h <- DESeq(dds_2h, parallel=TRUE)
save(dds_2h, file="../data/IFNa/RNA/diffAnalysis/dds_2h.rda")
res_2h <- results(dds_2h, independentFiltering=FALSE)
## 8 hours -----------------------------------
## Create DDS object and perform analysis
cts <- counts_8h$counts
colnames(cts) <- names_8h
coldata <- data.frame("treatment"=gsub("_[0-9]$", "", names_8h))
dds_8h <- DESeqDataSetFromMatrix(countData = cts,
colData = coldata,
design= ~ treatment)
## Run analysis in parallel
library("BiocParallel")
register(MulticoreParam(6))
dds_8h <- DESeq(dds_8h, parallel=TRUE)
save(dds_8h, file="../data/IFNa/RNA/diffAnalysis/dds_8h.rda")
res_8h <- results(dds_8h, independentFiltering=FALSE)
## 24hours -----------------------------------
## Create DDS object and perform analysis
cts <- counts_24h$counts
colnames(cts) <- names_24h
coldata <- data.frame("treatment"=gsub("_[0-9]$", "", names_24h))
dds_24h <- DESeqDataSetFromMatrix(countData = cts,
colData = coldata,
design= ~ treatment)
## Run analysis in parallel
library("BiocParallel")
register(MulticoreParam(6))
dds_24h <- DESeq(dds_24h, parallel=TRUE)
save(dds_24h, file="../data/IFNa/RNA/diffAnalysis/dds_24h.rda")
res_24h <- results(dds_24h, independentFiltering=FALSE)
###############################################################################
## 3) Parse results -----------------------------------------------------------
## 2 hours --------------------------
## Edit results
res.df_2h <- data.frame(res_2h)
counts_2h <- counts(dds_2h, normalized=TRUE)
counts_2h <- data.frame(counts_2h)
res.df_2h <- merge(res.df_2h, counts_2h, by="row.names")
colnames(res.df_2h)[1] <- "ensembl_gene_id"
## Add mean counts for conditions
res.df_2h$mean.ctrl <- apply(res.df_2h[,8:12], 1, mean)
res.df_2h$mean.ifna <- apply(res.df_2h[,13:17], 1, mean)
## Classify regions
res.df_2h$type <- "equal-regulated"
res.df_2h$type[res.df_2h$padj<=0.05 & res.df_2h$log2FoldChange>=1] <- "up-regulated"
res.df_2h$type[res.df_2h$padj<=0.05 & res.df_2h$log2FoldChange<=-1] <- "down-regulated"
res.df_2h$type[is.na(res.df_2h$padj)] <- "not-expressed"
## Add coordinates
ensembl = biomaRt::useEnsembl(biomart = "ensembl", GRCh = 37,
dataset = "hsapiens_gene_ensembl")
dat.hs = biomaRt::getBM(attributes = c("external_gene_name",
"ensembl_gene_id",
"chromosome_name",
"start_position",
"end_position"),
filters="ensembl_gene_id",
values = res.df_2h$ensembl_gene_id,
mart = ensembl)
res.df_2h <- merge(res.df_2h, dat.hs)
res.df_2h <- res.df_2h[res.df_2h$chromosome_name %in% c(1:22),]
res.df_2h$chromosome_name <- paste0("chr", res.df_2h$chromosome_name)
save(res.df_2h, file="../data/IFNa/RNA/diffAnalysis/res_2h.rda")
res_2h.gr <- regioneR::toGRanges(res.df_2h[,c(22:24,1,21,2:20)])
save(res_2h.gr, file="../data/IFNa/RNA/diffAnalysis/res_2h_granges.rda")
## 8 hours --------------------------
## Edit results
res.df_8h <- data.frame(res_8h)
counts_8h <- counts(dds_8h, normalized=TRUE)
counts_8h <- data.frame(counts_8h)
res.df_8h <- merge(res.df_8h, counts_8h, by="row.names")
colnames(res.df_8h)[1] <- "ensembl_gene_id"
## Add mean counts for conditions
res.df_8h$mean.ctrl <- apply(res.df_8h[,8:11], 1, mean)
res.df_8h$mean.ifna <- apply(res.df_8h[,12:16], 1, mean)
## Classify regions
res.df_8h$type <- "equal-regulated"
res.df_8h$type[res.df_8h$padj<=0.05 & res.df_8h$log2FoldChange>=1] <- "up-regulated"
res.df_8h$type[res.df_8h$padj<=0.05 & res.df_8h$log2FoldChange<=-1] <- "down-regulated"
res.df_8h$type[is.na(res.df_8h$padj)] <- "not-expressed"
## Add coordinates
ensembl = biomaRt::useEnsembl(biomart = "ensembl", GRCh = 37,
dataset = "hsapiens_gene_ensembl")
dat.hs = biomaRt::getBM(attributes = c("external_gene_name",
"ensembl_gene_id",
"chromosome_name",
"start_position",
"end_position"),
filters="ensembl_gene_id",
values = res.df_8h$ensembl_gene_id,
mart = ensembl)
res.df_8h <- merge(res.df_8h, dat.hs)
res.df_8h <- res.df_8h[res.df_8h$chromosome_name %in% c(1:22),]
res.df_8h$chromosome_name <- paste0("chr", res.df_8h$chromosome_name)
save(res.df_8h, file="../data/IFNa/RNA/diffAnalysis/res_8h.rda")
res_8h.gr <- regioneR::toGRanges(res.df_8h[,c(21:23,1,20,2:19)])
save(res_8h.gr, file="../data/IFNa/RNA/diffAnalysis/res_8h_granges.rda")
## 24 hours -------------------------
## Edit results
res.df_24h <- data.frame(res_24h)
counts_24h <- counts(dds_24h, normalized=TRUE)
counts_24h <- data.frame(counts_24h)
res.df_24h <- merge(res.df_24h, counts_24h, by="row.names")
colnames(res.df_24h)[1] <- "ensembl_gene_id"
## Add mean counts for conditions
res.df_24h$mean.ctrl <- apply(res.df_24h[,8:12], 1, mean)
res.df_24h$mean.ifna <- apply(res.df_24h[,13:17], 1, mean)
## Classify regions
res.df_24h$type <- "equal-regulated"
res.df_24h$type[res.df_24h$padj<=0.05 & res.df_24h$log2FoldChange>=1] <- "up-regulated"
res.df_24h$type[res.df_24h$padj<=0.05 & res.df_24h$log2FoldChange<=-1] <- "down-regulated"
res.df_24h$type[is.na(res.df_24h$padj)] <- "not-expressed"
## Add coordinates
ensembl = biomaRt::useEnsembl(biomart = "ensembl", GRCh = 37,
dataset = "hsapiens_gene_ensembl")
dat.hs = biomaRt::getBM(attributes = c("external_gene_name",
"ensembl_gene_id",
"chromosome_name",
"start_position",
"end_position"),
filters="ensembl_gene_id",
values = res.df_24h$ensembl_gene_id,
mart = ensembl)
res.df_24h <- merge(res.df_24h, dat.hs)
res.df_24h <- res.df_24h[res.df_24h$chromosome_name %in% c(1:22),]
res.df_24h$chromosome_name <- paste0("chr", res.df_24h$chromosome_name)
save(res.df_24h, file="../data/IFNa/RNA/diffAnalysis/res_24h.rda")
res_24h.gr <- regioneR::toGRanges(res.df_24h[,c(22:24,1,21,2:20)])
save(res_24h.gr, file="../data/IFNa/RNA/diffAnalysis/res_24h_granges.rda")Correlation between OCRs and gene expression
Annotate OCRs to genes
As OCRs can regulate more than one gene and a single gene can be regulated by several OCRs, to associate genes and OCRs we used a 40kb window. This means that a gene i is associated to all the OCRs that overlap with a 40kb window centered on the gene’s TSS.
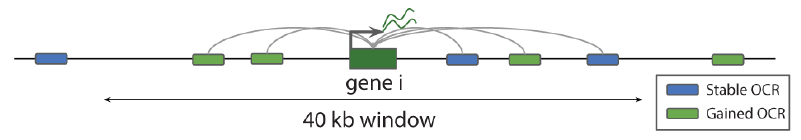
## Load genes and create windows
load("~/data/genesCoding_gencodev18_granges.rda")
win=40000
genes.prom <- promoters(genes, upstream=win/2, downstream=win/2)
## Load OCRs 2h
load("../data/IFNa/ATAC/diffAnalysis/res_2h_granges.rda")
ols <- findOverlaps(res.gr, genes.prom)
ols_df <- cbind(data.frame(res.gr)[queryHits(ols), c(6,8,13)],
data.frame(genes)[subjectHits(ols), c(6,7)])
ols_split <- split(ols_df, ols_df$GeneID)
ols_final <- do.call(rbind,
lapply(ols_split,
function(x) data.frame(GeneID=unique(x$GeneID),
ensembl_gene_id=paste0(unique(x$ensembl_gene_id),
collapse=", "),
gene_name=paste0(unique(x$external_gene_name),
collapse=", "),
stringsAsFactors=FALSE)))
coords <- data.frame("Coordinates"=as.character(res.gr),
"GeneID"=res.gr$GeneID,
"log2FoldChange"=res.gr$log2FoldChange,
"OCR type"=res.gr$type,
stringsAsFactors=FALSE)
anno <- dplyr::left_join(coords, ols_final)
anno$ensembl_gene_id[is.na(anno$ensembl_gene_id)] <- "None"
anno$gene_name[is.na(anno$gene_name)] <- "None"
save(anno, file=file.path(out_dir, "ATAC_annotation_40kb_coding_2h.rda"))
## Load OCRs 24h
load("../data/IFNa/ATAC/diffAnalysis/res_24h_granges.rda")
ols <- findOverlaps(res.gr, genes.prom)
ols_df <- cbind(data.frame(res.gr)[queryHits(ols), c(6,8,13)],
data.frame(genes)[subjectHits(ols), c(6,7)])
ols_split <- split(ols_df, ols_df$GeneID)
ols_final <- do.call(rbind,
lapply(ols_split,
function(x) data.frame(GeneID=unique(x$GeneID),
ensembl_gene_id=paste0(unique(x$ensembl_gene_id),
collapse=", "),
gene_name=paste0(unique(x$external_gene_name),
collapse=", "),
stringsAsFactors=FALSE)))
coords <- data.frame("Coordinates"=as.character(res.gr),
"GeneID"=res.gr$GeneID,
"log2FoldChange"=res.gr$log2FoldChange,
"OCR type"=res.gr$type,
stringsAsFactors=FALSE)
anno <- dplyr::left_join(coords, ols_final)
anno$ensembl_gene_id[is.na(anno$ensembl_gene_id)] <- "None"
anno$gene_name[is.na(anno$gene_name)] <- "None"
save(anno, file=file.path(out_dir, "ATAC_annotation_40kb_coding_24h.rda"))The following tables contain a list of all the gained OCRs and the genes to which they have been annotated. For a full list of OCRs please go to the original publication Supplementary Data 2 (Download)
load(file.path(out_dir, "ATAC_annotation_40kb_coding_2h.rda"))
DT::datatable(anno[anno$OCR_type=="gained",],
rownames=FALSE,
caption = "Gained OCRs at 2 hours and the genes to which they are annotated, using a 40kb window.")load(file.path(out_dir, "ATAC_annotation_40kb_coding_24h.rda"))
DT::datatable(anno[anno$OCR_type=="gained",],
rownames=FALSE,
caption = "Gained OCRs at 24 hours and the genes to which they are annotated, using a 40kb window.")Next, we intersect the OCRs annotated to genes with the RNA-seq data in the different time points (2, 8 and 24 hours) generated in the section above.
load(file.path(out_dir, "ATAC_annotation_40kb_coding_2h.rda"))
colnames(anno)[6] <- "OCR_type"
anno$time <- "ATAC 2 h"
anno_all <- anno
load(file.path(out_dir, "ATAC_annotation_40kb_coding_24h.rda"))
colnames(anno)[6] <- "OCR_type"
anno$time <- "ATAC 24 h"
anno_all <- rbind(anno_all, anno)
anno_all <- anno_all[,c("Region_ID", "OCR_type", "ensembl_gene_id", "time")]
## Separate ensmbl ids
ids <- strsplit(anno_all$ensembl_gene_id, ", ")
len <- sapply(ids, length)
anno_all <- anno_all[unlist(mapply(rep, x=1:nrow(anno_all), each=len)),]
anno_all$ensembl_gene_id <- unlist(ids)
anno_all <- anno_all[anno_all$ensembl_gene_id!="None",]
## Load gene expression data
load("../data/IFNa/RNA/diffAnalysis/res_2h.rda")
res_2h <- res.df_2h[,c("ensembl_gene_id", "log2FoldChange", "type")]
colnames(res_2h)[2:3] <- paste0("RNA_", colnames(res_2h)[2:3])
res_2h$RNA_time <- "RNA 2 h"
load("../data/IFNa/RNA/diffAnalysis/res_8h.rda")
res_8h <- res.df_8h[,c("ensembl_gene_id", "log2FoldChange", "type")]
colnames(res_8h)[2:3] <- paste0("RNA_", colnames(res_8h)[2:3])
res_8h$RNA_time <- "RNA 8 h"
load("../data/IFNa/RNA/diffAnalysis/res_24h.rda")
res_24h <- res.df_24h[,c("ensembl_gene_id", "log2FoldChange", "type")]
colnames(res_24h)[2:3] <- paste0("RNA_", colnames(res_24h)[2:3])
res_24h$RNA_time <- "RNA 24 h"
rna_all <- rbind(res_2h, res_8h, res_24h)
save(rna_all, file=file.path(out_dir, "RNA_merged_datasets.rda"))
## Intersect RNA-seq with ATAC-seq data
anno_all <- dplyr::left_join(anno_all, rna_all)
anno_all$time <- factor(anno_all$time, levels=c("ATAC 2 h", "ATAC 24 h"))
anno_all$RNA_time <- factor(anno_all$RNA_time, levels=c("RNA 2 h", "RNA 8 h", "RNA 24 h"))
anno_all$OCR_type <- factor(anno_all$OCR_type, levels=c("stable", "gained", "lost"))
save(anno_all, file=file.path(out_dir, "ATAC-RNA_intersection_40kb.rda"))Correlation with gene expression
load(file.path(out_dir, "ATAC-RNA_intersection_40kb.rda"))
ggplot(anno_all[anno_all$OCR_type!="lost",],
aes(OCR_type, ..count.., fill=RNA_type)) +
geom_bar(position="fill", color="black", lwd=0.7) +
scale_fill_manual(values=pals$diff_rna,
name="RNA type") +
facet_grid(RNA_time~time) +
theme(legend.position="top") +
xlab("OCR type") + ylab("Proportion") +
guides(fill=guide_legend(nrow=2, byrow=T))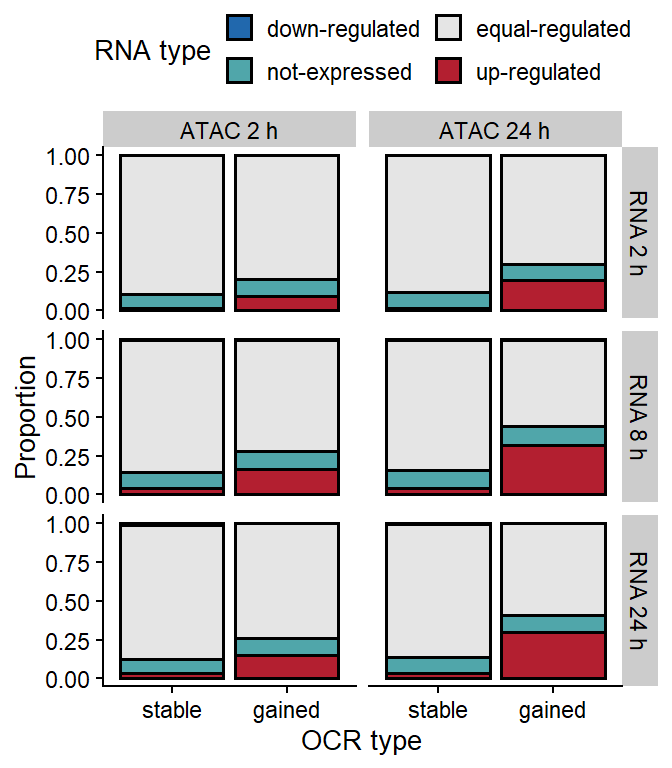
Figure 1: Frequency of upregulated, downregulated or stable transcripts in the vicinity (<20 kb transcription start site (TSS) distance) of differnt types of OCRs.
To see the if there exists and additive effect of OCRs regulating a specific transcript, we group genes by number of gained or stable OCRs in the vicinity.
load(file.path(out_dir, "ATAC-RNA_intersection_40kb.rda"))
counts <- anno_all %>%
filter(OCR_type != "lost") %>%
group_by(RNA_time, ensembl_gene_id, RNA_log2FoldChange, RNA_type, time, OCR_type) %>%
summarise(num = n())
ids_no_gained <- counts$ensembl_gene_id[!(unique(counts$ensembl_gene_id[counts$OCR_type=="stable"]) %in% unique(counts$ensembl_gene_id[counts$OCR_type=="gained"]))]
ids_no_stable <- counts$ensembl_gene_id[!(unique(counts$ensembl_gene_id[counts$OCR_type=="gained"]) %in% unique(counts$ensembl_gene_id[counts$OCR_type=="stable"]))]
## Add genes with 0 gained OCRs but stable OCRs
rep <- counts[which(counts$ensembl_gene_id %in% ids_no_gained),]
rep$OCR_type <- "gained"
rep$num <- 0
counts <- rbind(counts, rep)
## Add genes with 0 stable OCRs but gained OCRs
rep <- counts[which(counts$ensembl_gene_id %in% ids_no_stable),]
rep$OCR_type <- "stable"
rep$num <- 0
counts <- rbind(counts, rep)
## Add genes with 0 OCRs
load(file.path(out_dir, "RNA_merged_datasets.rda"))
no_ocr <- rna_all[!(rna_all$ensembl_gene_id %in% unique(counts$ensembl_gene_id)),
c("RNA_time", "ensembl_gene_id", "RNA_log2FoldChange", "RNA_type")]
len <- nrow(no_ocr)
no_ocr <- rbind(no_ocr, no_ocr, no_ocr, no_ocr)
no_ocr$time <- rep(c("ATAC 2 h", "ATAC 24 h"), each=len*2)
no_ocr$OCR_type <- rep(c("stable", "gained", "stable", "gained"), each=len)
counts <- rbind(counts, no_ocr)
## Group according to number of OCRs
counts$group <- "0"
counts$group[counts$num==1] <- "1"
counts$group[counts$num>1] <- ">1"
counts$group <- factor(counts$group, levels=c("0", "1", ">1"))
counts$OCR_type <- factor(counts$OCR_type, levels=c("stable", "gained"))
ggplot(counts, aes(group, ..count.., fill=RNA_type)) +
geom_bar(position="fill", color="black", lwd=0.7) +
scale_fill_manual(values=pals$diff_rna,
name="RNA Type") +
theme(axis.text.x=element_text(angle=30, hjust=1),
legend.position = "top") +
guides(fill=guide_legend(nrow=2, byrow=T)) +
xlab("# OCRs of same type") +
ylab("Proportion") +
facet_grid(RNA_time~time+OCR_type)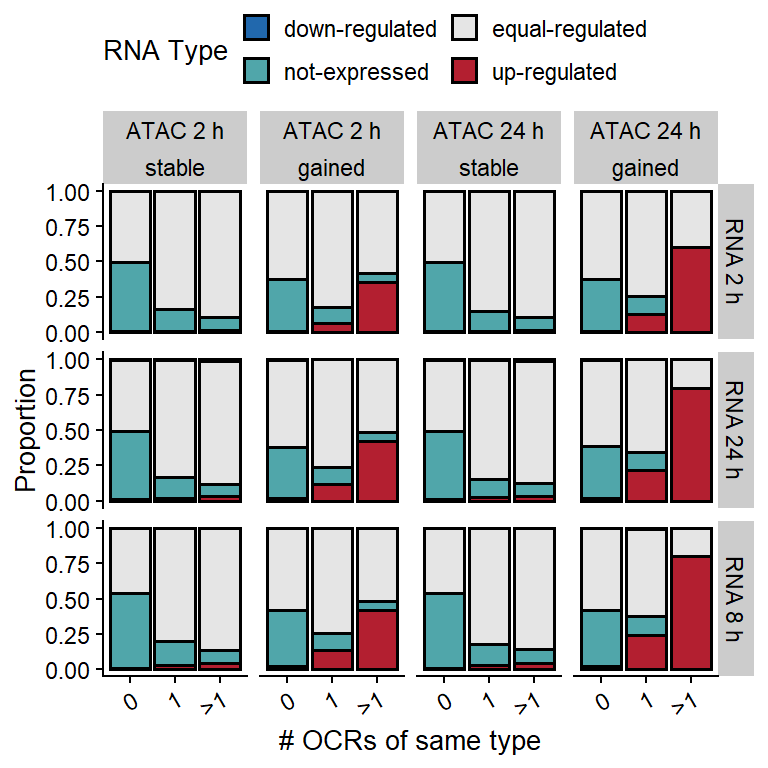
Figure 2: Frequency of transcripts of each type classified by the number gained or stable OCRs located in the vicinity, at different time points.
## Plot results
ggplot(counts,
aes(group, RNA_log2FoldChange)) +
geom_hline(yintercept=0, lty=2, color="grey") +
geom_boxplot(outlier.shape=NA, lwd=0.7) +
coord_cartesian(ylim=c(-1, 11)) +
xlab("# OCRs of same type") +
ylab("RNA-seq log2 Fold-Change") +
facet_grid(RNA_time~time+OCR_type)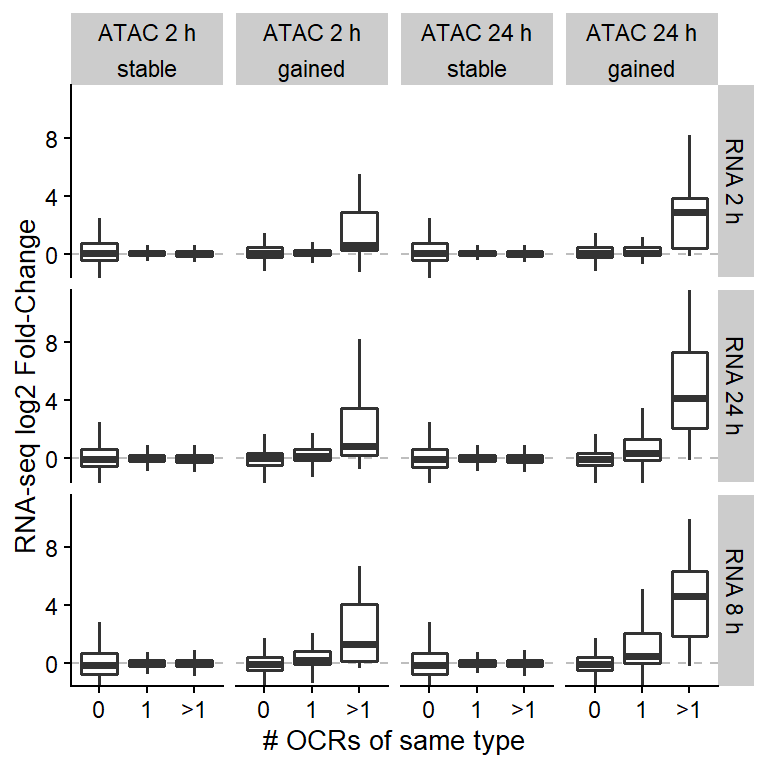
Figure 3: mRNA log2 fold-changes detected by RNA-seq are related to the type and the number of OCRs.
sessionInfo()R version 4.0.2 (2020-06-22)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 18362)
Matrix products: default
locale:
[1] LC_COLLATE=Spanish_Spain.1252 LC_CTYPE=Spanish_Spain.1252
[3] LC_MONETARY=Spanish_Spain.1252 LC_NUMERIC=C
[5] LC_TIME=Spanish_Spain.1252
attached base packages:
[1] grid parallel stats4 stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] DESeq2_1.29.7 SummarizedExperiment_1.19.6
[3] DelayedArray_0.15.7 matrixStats_0.56.0
[5] Matrix_1.2-18 Biobase_2.49.0
[7] ComplexHeatmap_2.5.3 dplyr_1.0.0
[9] kableExtra_1.1.0 cowplot_1.0.0
[11] ggplot2_3.3.2 GenomicRanges_1.41.5
[13] GenomeInfoDb_1.25.8 IRanges_2.23.10
[15] S4Vectors_0.27.12 BiocGenerics_0.35.4
[17] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] bitops_1.0-6 fs_1.4.2 bit64_0.9-7.1
[4] webshot_0.5.2 RColorBrewer_1.1-2 httr_1.4.2
[7] rprojroot_1.3-2 tools_4.0.0 backports_1.1.8
[10] DT_0.14 R6_2.4.1 DBI_1.1.0
[13] colorspace_1.4-1 GetoptLong_1.0.2 withr_2.2.0
[16] tidyselect_1.1.0 bit_1.1-15.2 compiler_4.0.0
[19] git2r_0.27.1 rvest_0.3.6 xml2_1.3.2
[22] labeling_0.3 bookdown_0.20 scales_1.1.1
[25] readr_1.3.1 genefilter_1.71.0 stringr_1.4.0
[28] digest_0.6.25 rmarkdown_2.3 XVector_0.29.3
[31] pkgconfig_2.0.3 htmltools_0.5.0 highr_0.8
[34] htmlwidgets_1.5.1 rlang_0.4.7 GlobalOptions_0.1.2
[37] rstudioapi_0.11 RSQLite_2.2.0 farver_2.0.3
[40] shape_1.4.4 generics_0.0.2 jsonlite_1.7.0
[43] crosstalk_1.1.0.1 BiocParallel_1.23.2 RCurl_1.98-1.2
[46] magrittr_1.5 GenomeInfoDbData_1.2.3 Rcpp_1.0.5
[49] munsell_0.5.0 lifecycle_0.2.0 stringi_1.4.6
[52] icon_0.1.0 yaml_2.2.1 zlibbioc_1.35.0
[55] blob_1.2.1 promises_1.1.1 crayon_1.3.4
[58] lattice_0.20-41 splines_4.0.0 annotate_1.67.0
[61] circlize_0.4.10 hms_0.5.3 locfit_1.5-9.4
[64] knitr_1.29 pillar_1.4.6 rjson_0.2.20
[67] geneplotter_1.67.0 XML_3.99-0.5 glue_1.4.1
[70] evaluate_0.14 png_0.1-7 vctrs_0.3.2
[73] httpuv_1.5.4 gtable_0.3.0 purrr_0.3.4
[76] clue_0.3-57 xfun_0.16 xtable_1.8-4
[79] later_1.1.0.1 survival_3.2-3 viridisLite_0.3.0
[82] tibble_3.0.3 AnnotationDbi_1.51.3 memoise_1.1.0
[85] cluster_2.1.0 ellipsis_0.3.1