#4 Changes in 3D chormatin structure
Mireia Ramos-Rodríguez
Details
- Original publication:
Ramos-Rodríguez, M., Raurell-Vila, H., Colli, M.L. et al. The impact of proinflammatory cytokines on the β-cell regulatory landscape provides insights into the genetics of type 1 diabetes. Nat Genet. 51, 1588–1595 (2019) https://doi.org/10.1038/s41588-019-0524-6
Contents: Analyses and figures contained in this document correspond to the following figures/sections of the original publication:
- Results: “Changes in islet 3D chromatin structure”.
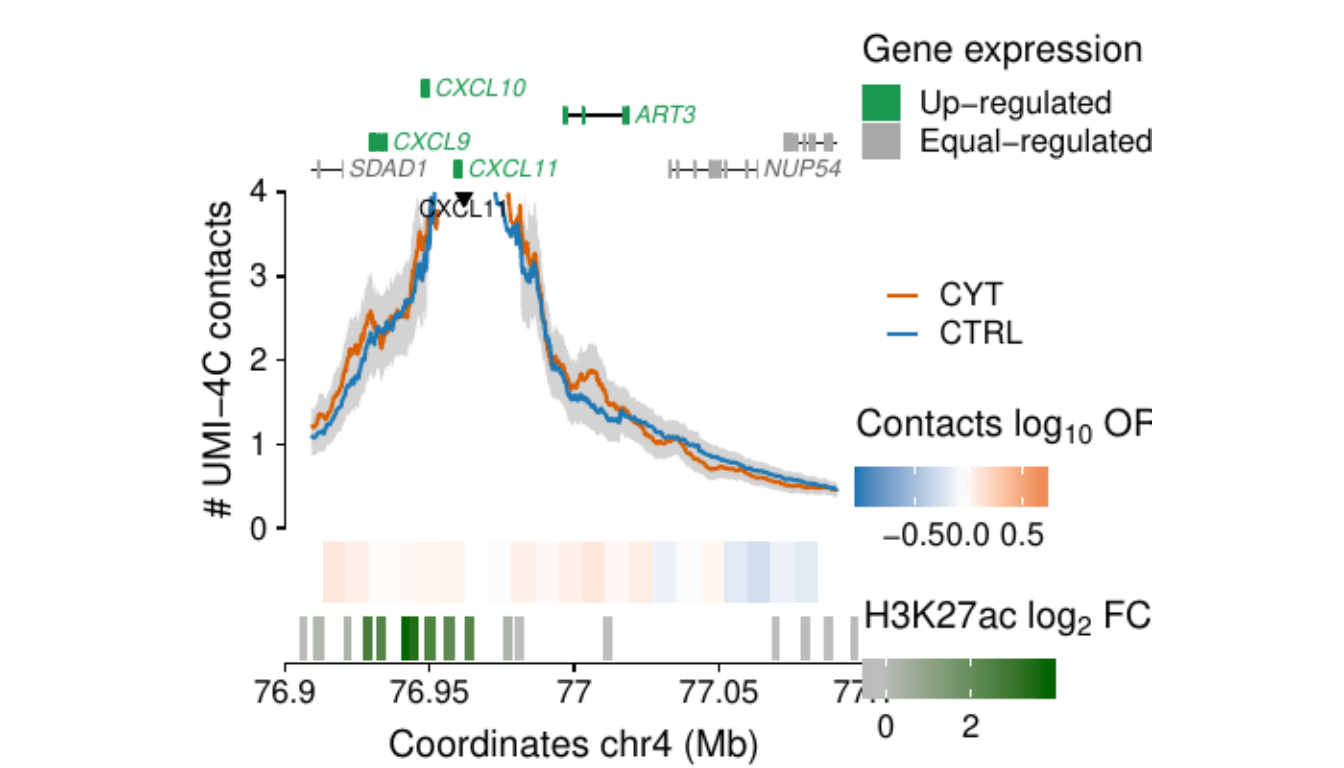
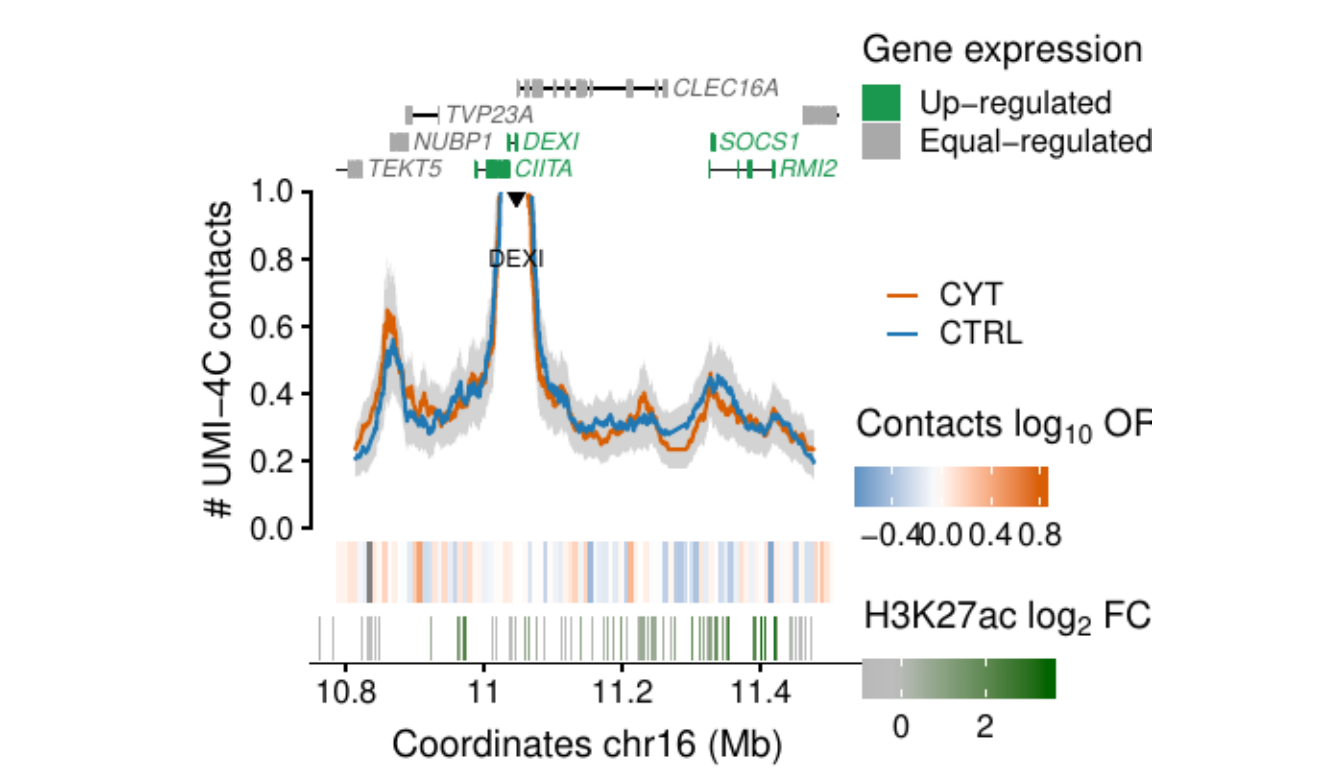
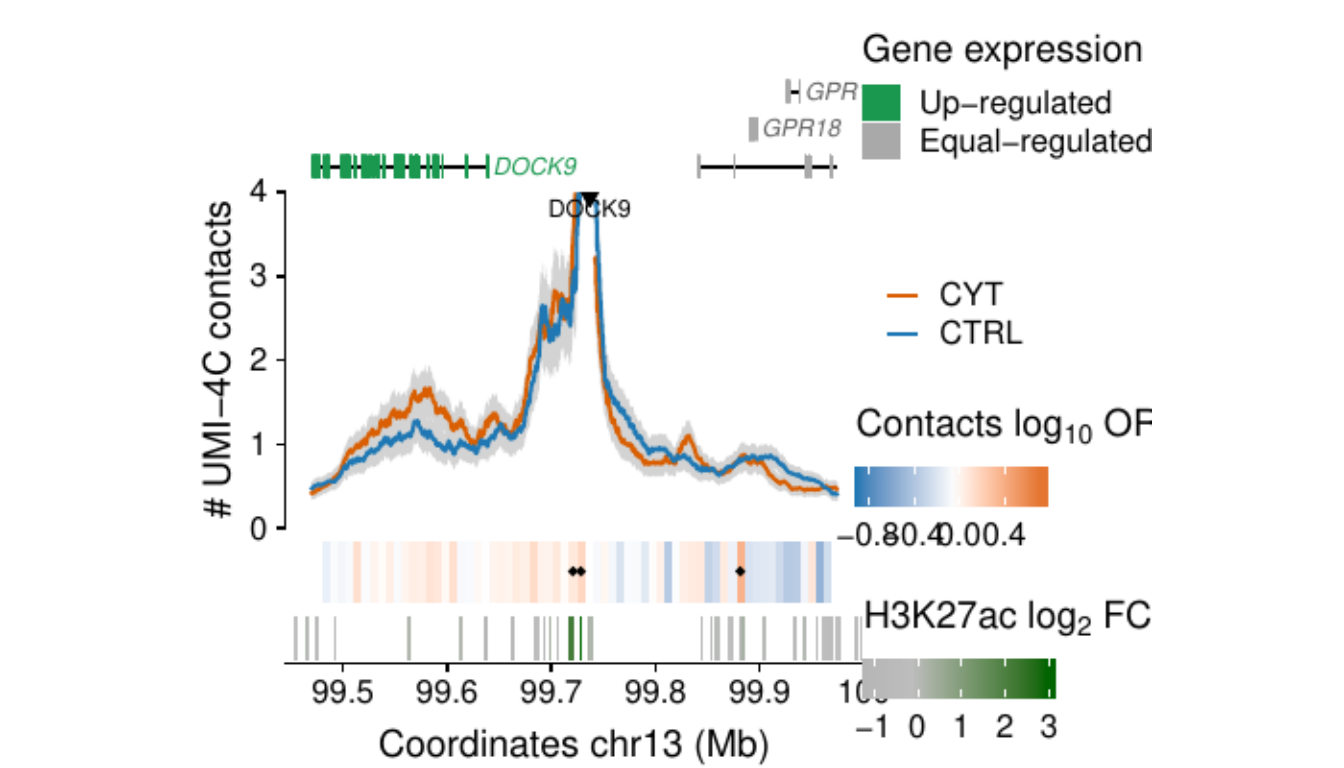
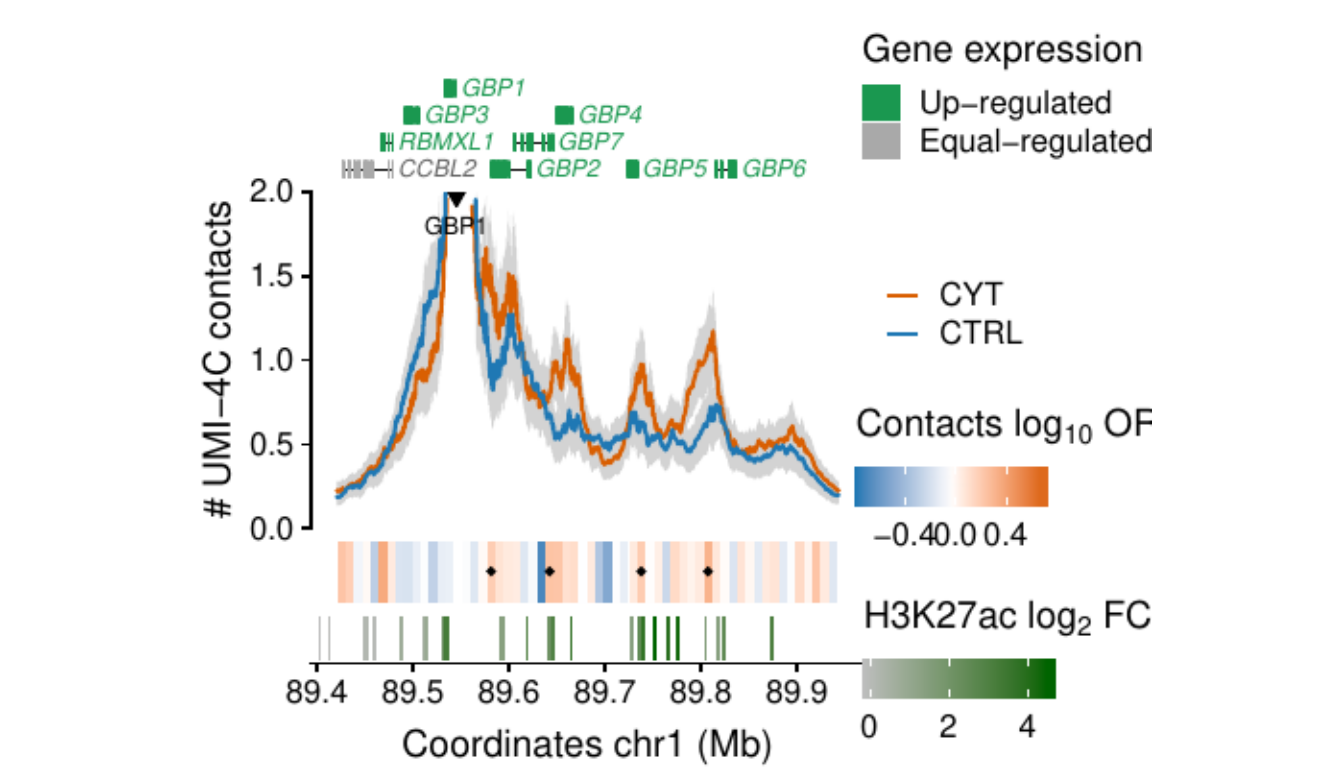
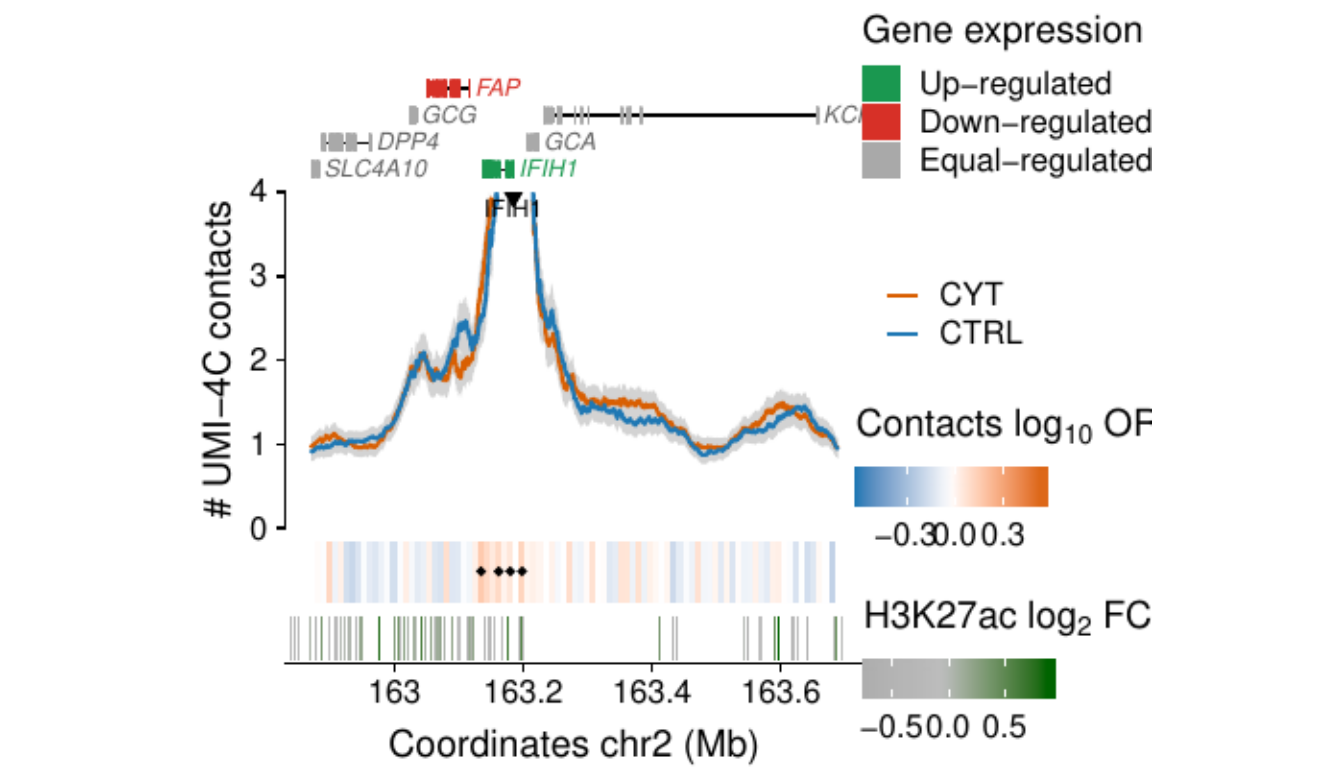
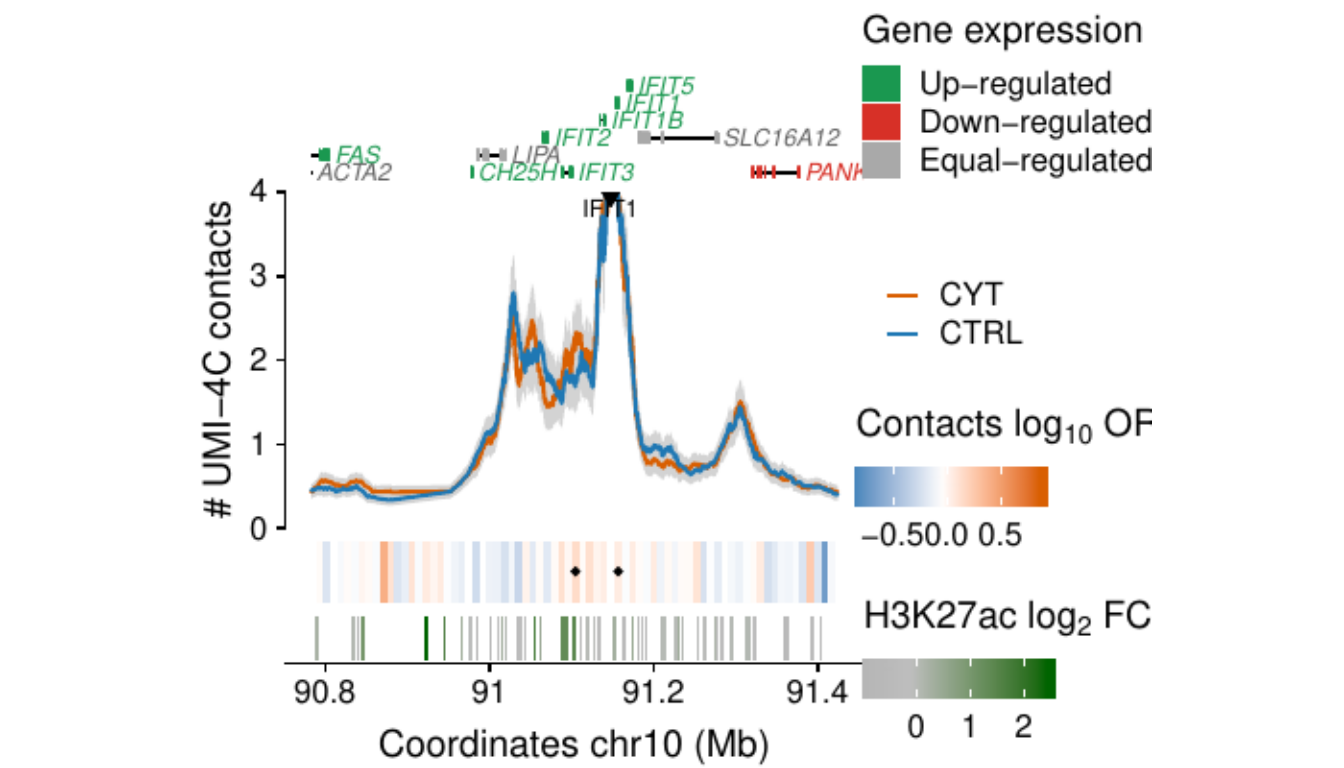
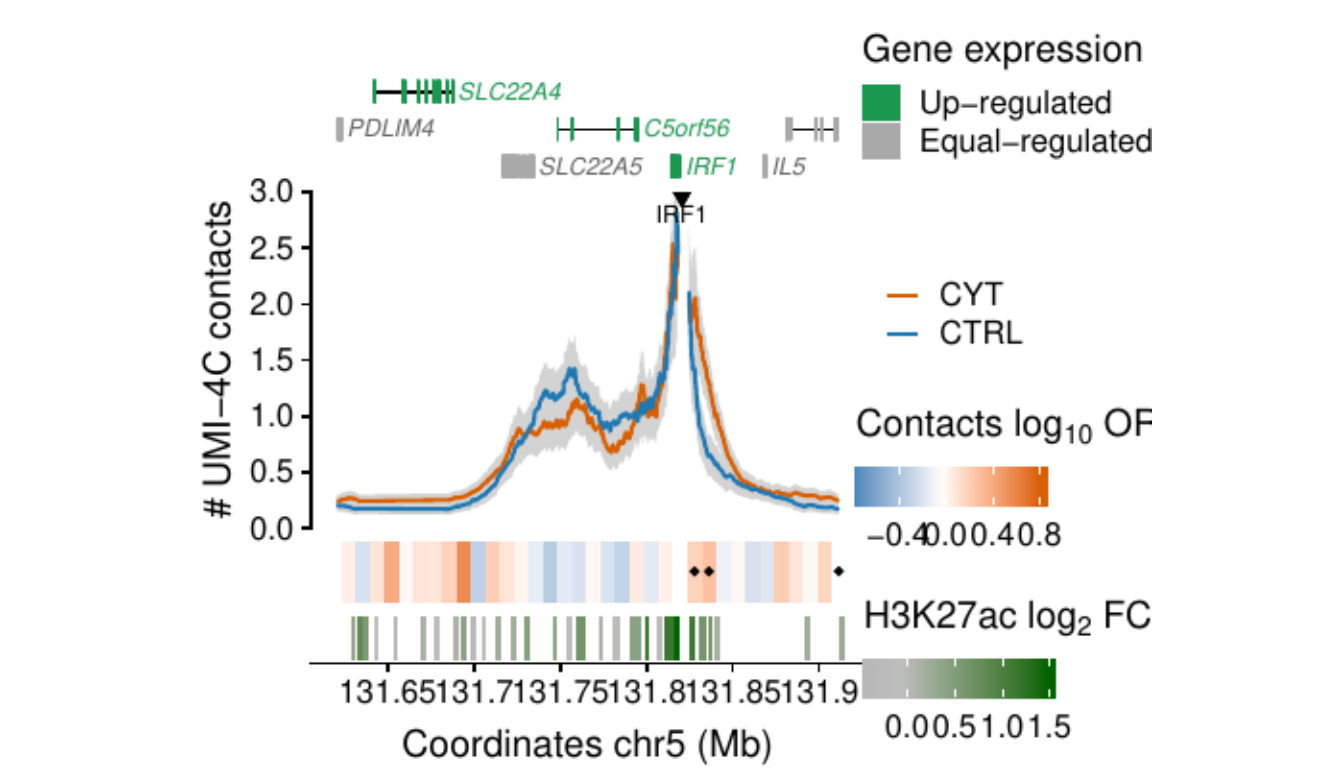
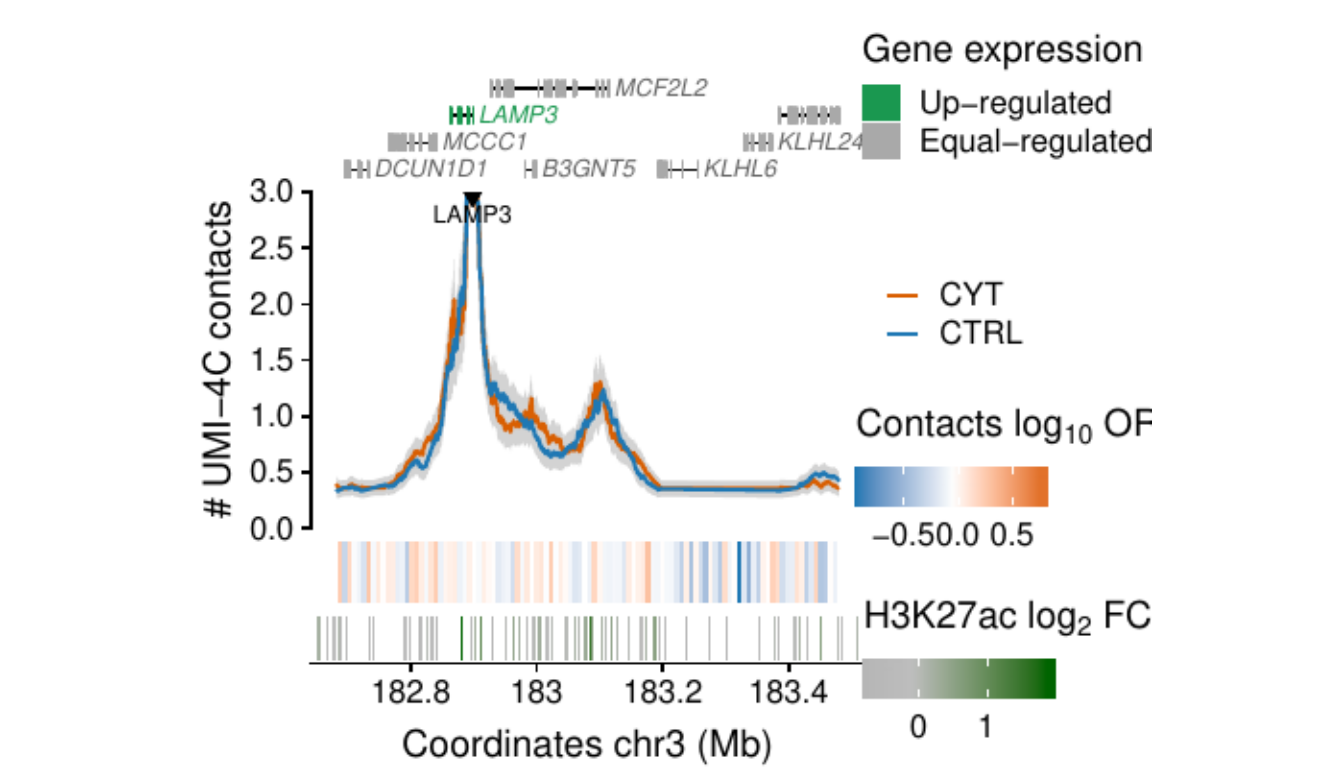
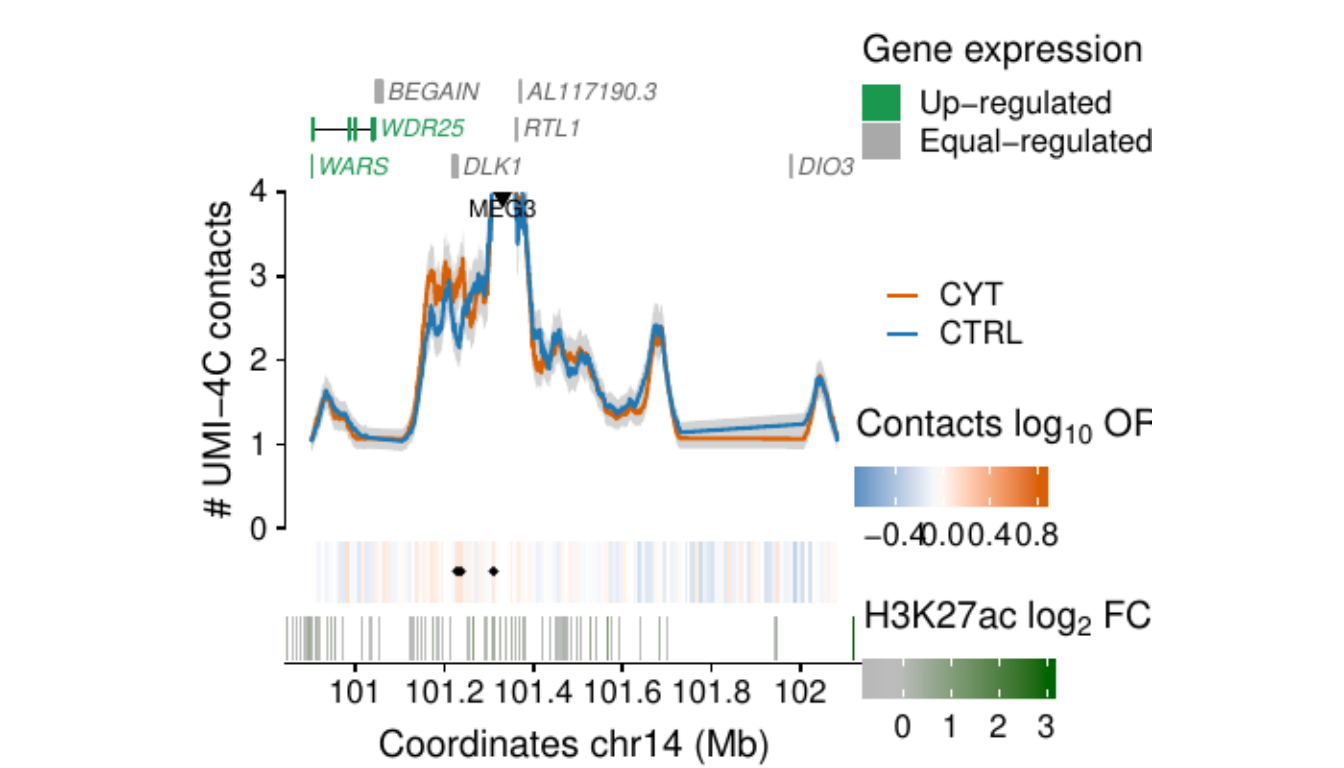
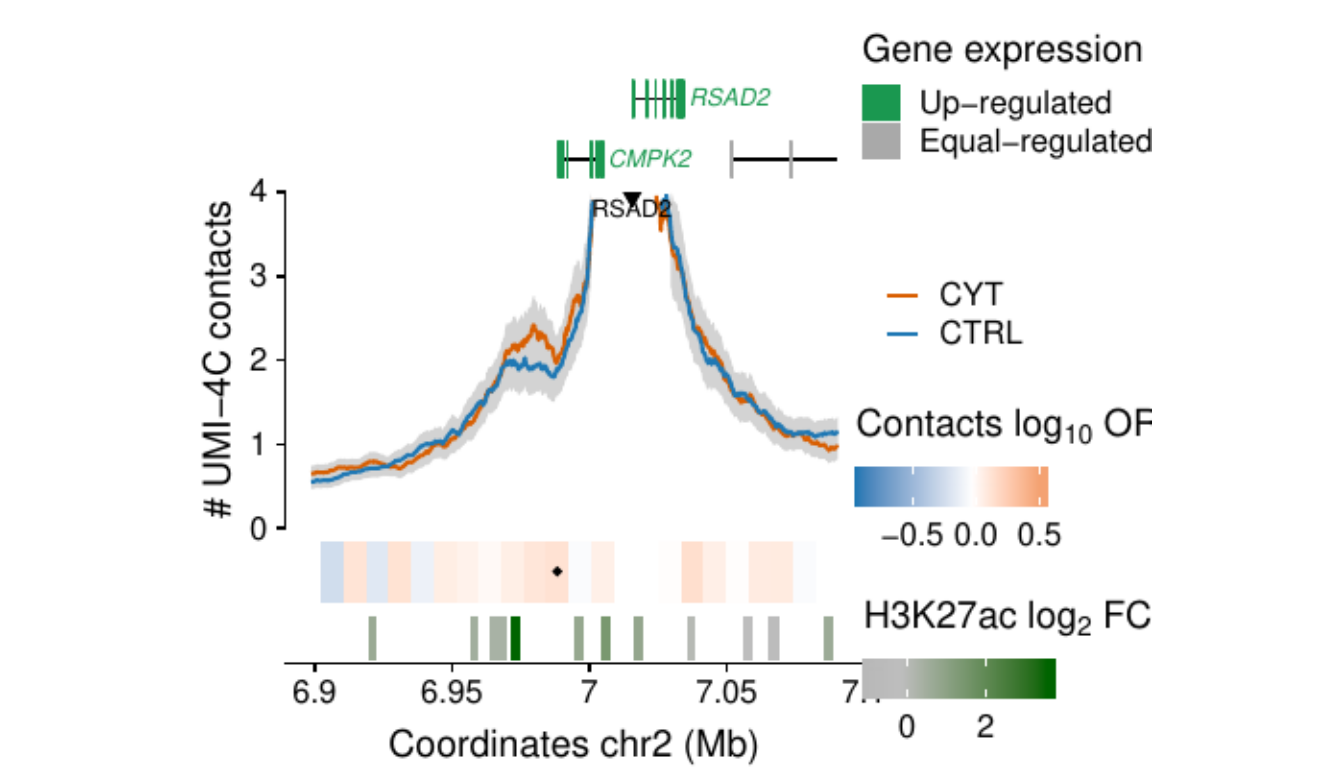
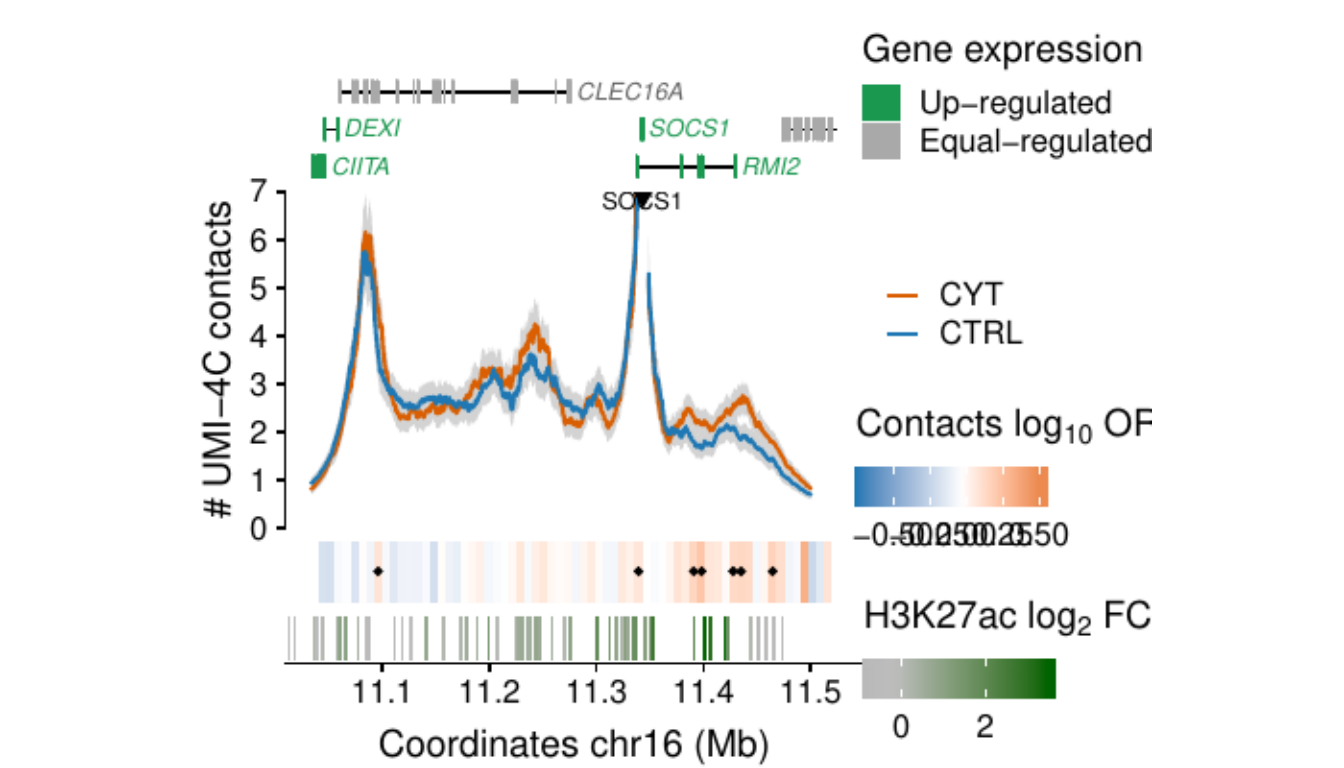
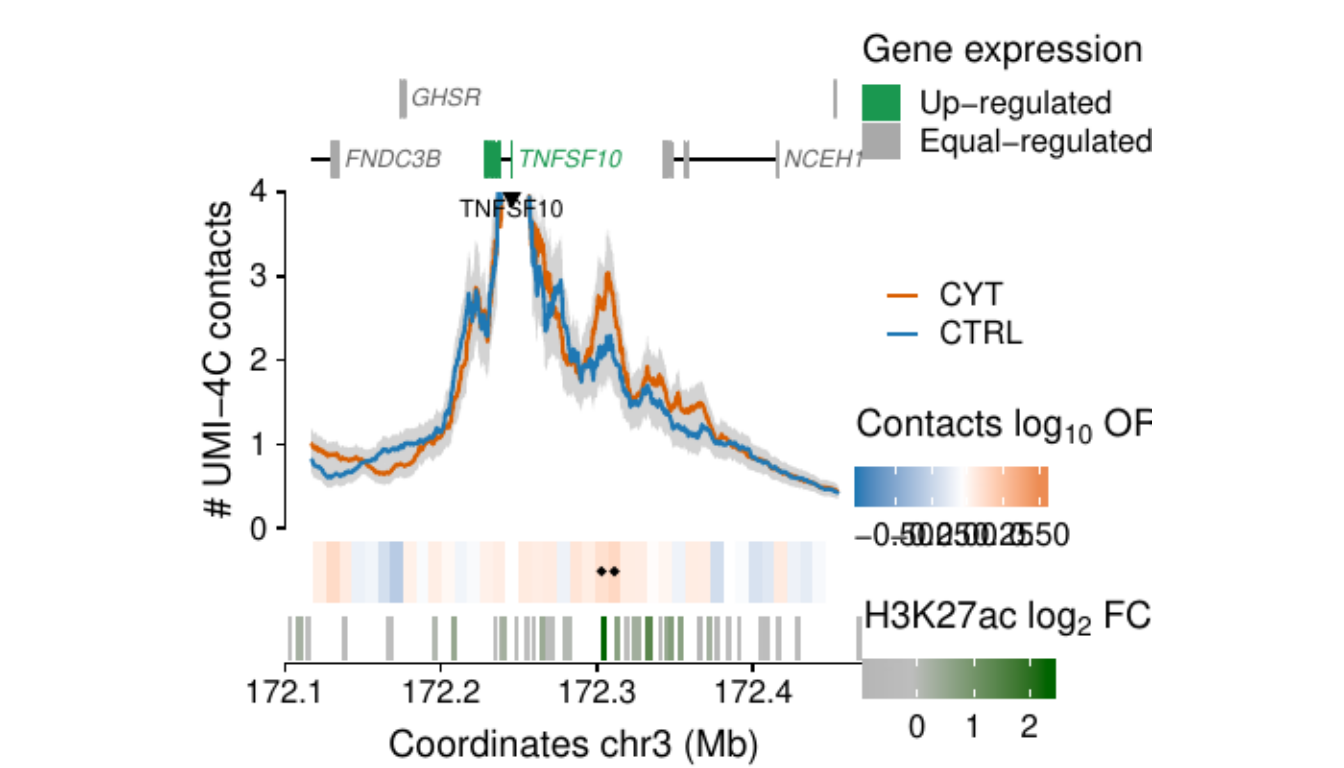
- Figure 3: “Cytokine exposure induces changes in human islet 3D chromatin structure”. Panels a to c.
- Extended Data Figure 5: “3D chromatin changes induced by exposure of human islets to pro-inflammatory cytokines”. Panels b to d.
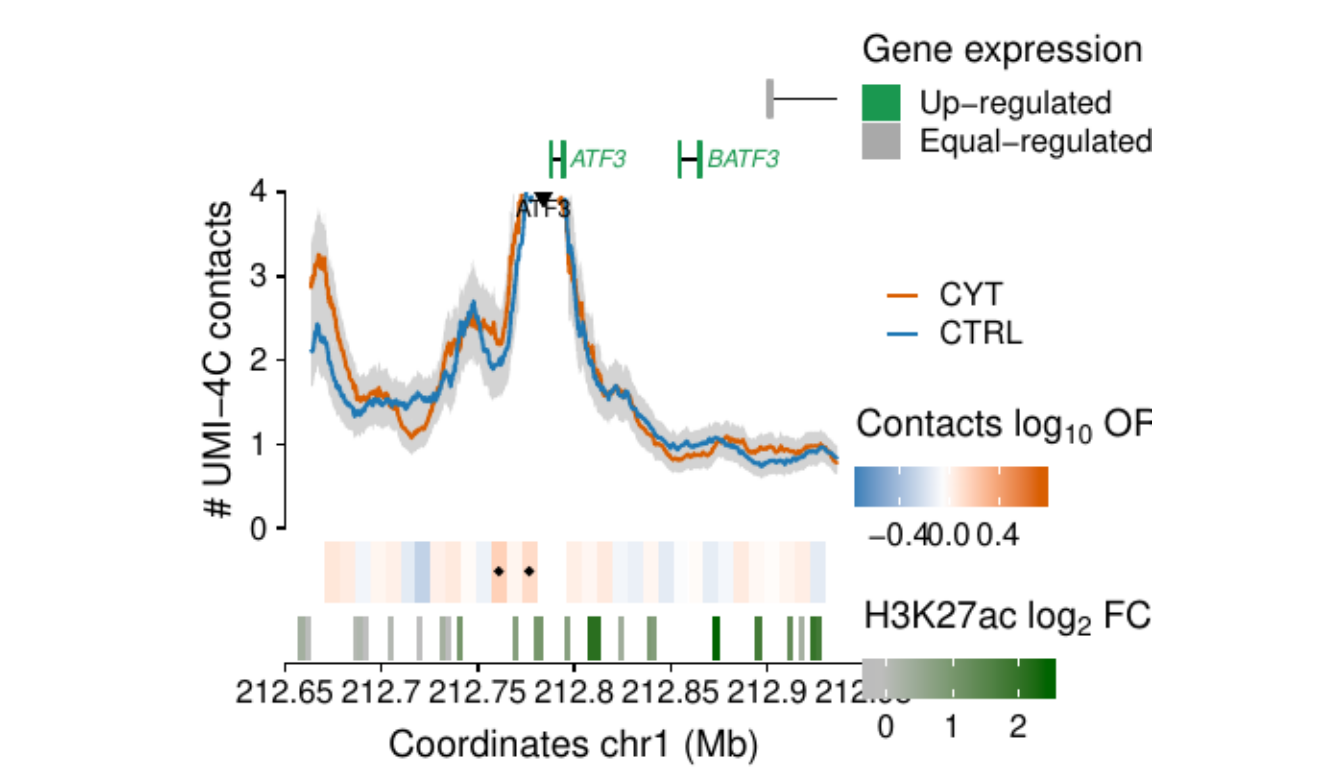
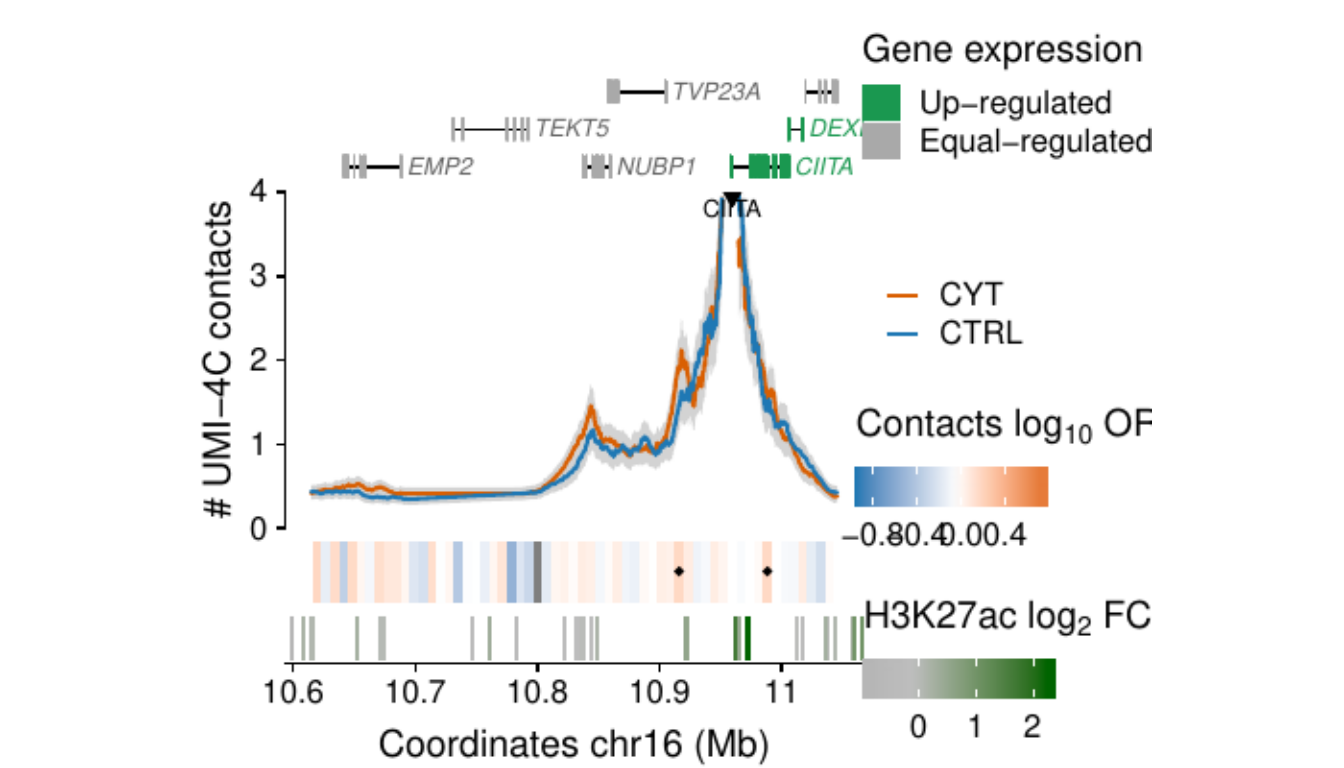
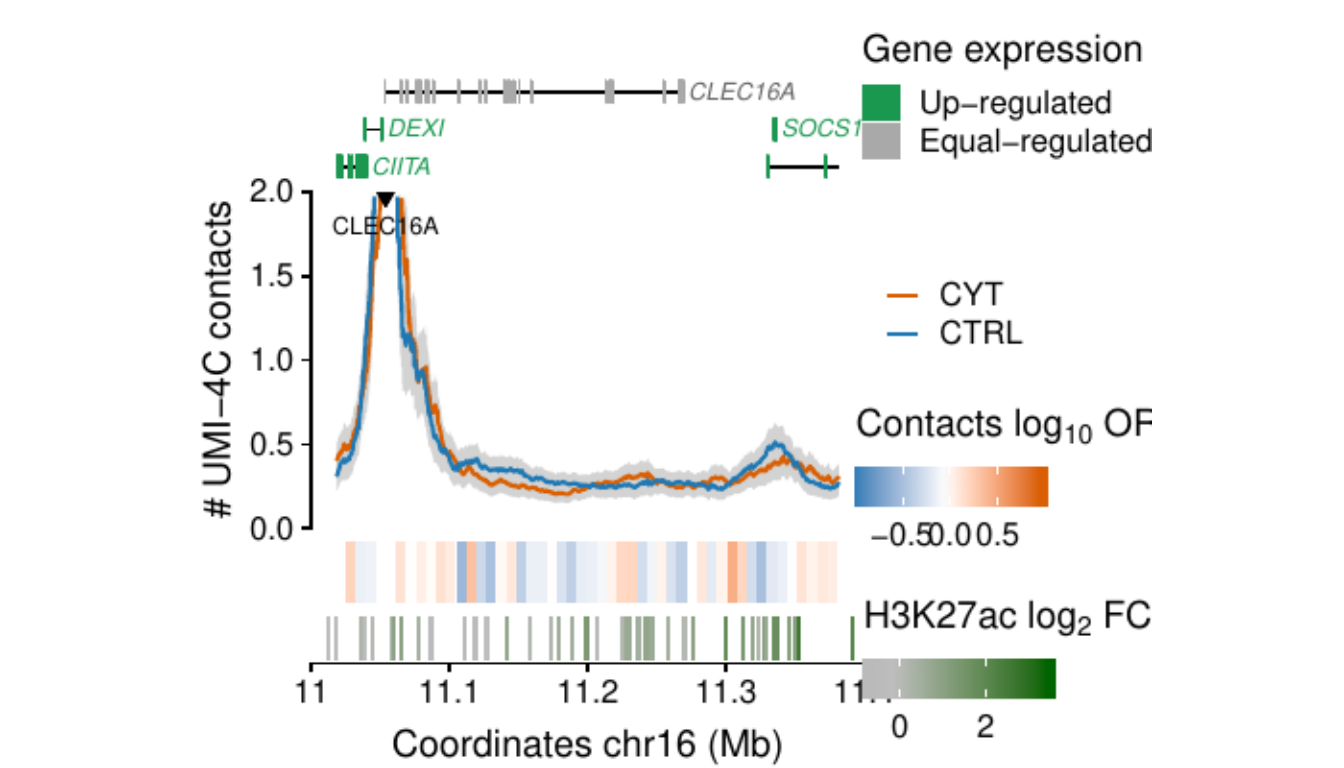
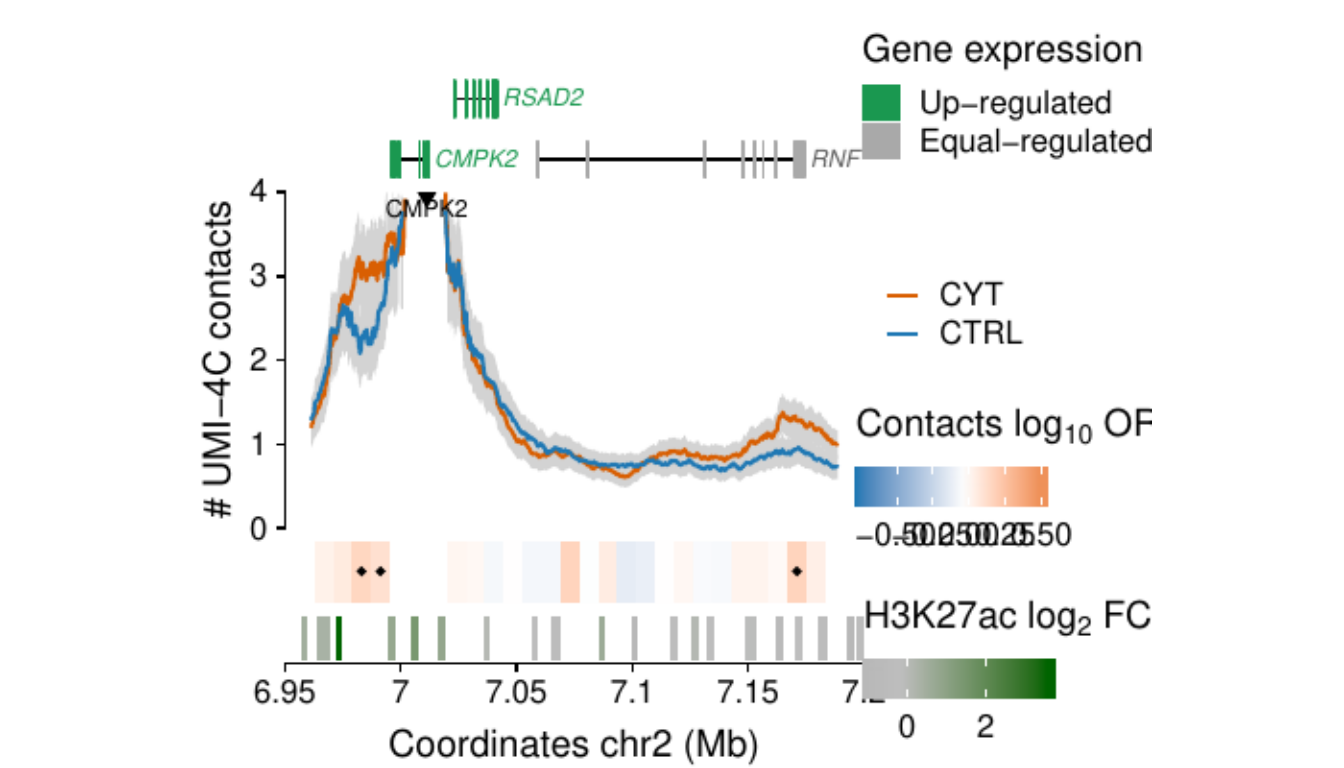
Process and calculate differential contacts at regulatory elements
Rscript code/CYT_UMI4C_processing.Rdevtools::load_all("~/tools/umi4cCatcheR/")
library(umi4cPackage)
conf <- "../data/CYT/UMI4C/conf/"
umi4cPackage::p4cLoadConfFiles(conf)
df <- read.delim("../data/CYT/UMI4C/UMI4C_promoters_views.tsv", stringsAsFactors=T, header=T)
tracks <- gtrack.ls()
for (i in 1:nrow(df)) {
sel <- tracks[grepl(df$bait[i], tracks) &
grepl("_m_", tracks)]
res <- process4CProfiles(treat_name=sel[2],
ctrl_name=sel[1],
scope=1e6,
min_win_mols=50,
name_bait=df$bait[i])
diff <- diffContacts(res,
times_mean=20,
exclude_viewpoint=3e3,
adj.threshold=0.05,
adj.method="none",
min_mols_test=0,
min_odds_ratio=1)
save(res, diff,
file=file.path(out_dir"UMI4C_norm_results_", df$bait[i], ".rda"))
}Plot UMI-4C at up-regulated gene promoters
## RE ---------------------------------------------------------------
load("../data/CYT/REs/REs_hi_fc1_padj0.05_granges_subgroup.rda")
## Genes ------------------------------------------------------------
load("../data/CYT//RNA/diffAnalysis/RNA_hi_GRangesBatch.rda")
res.gr$type[res.gr$baseMean<=1] <- "not-expressed"
res.gr <- res.gr[res.gr$gene_biotype=="protein_coding",]
col.df <- data.frame("type"=c(names(pals$differential), "not-expressed"),
"color"=c(pals$differential, "black"),
stringsAsFactors = FALSE)
col.df$color[grep("grey", col.df$color)] <- "grey39"
mcols(res.gr) <- dplyr::left_join(data.frame(mcols(res.gr)[,c(1:2,10)]),
col.df)
colnames(mcols(res.gr))[1] <- "ensembl_gene_id"
mcol.color=4
mcol.name=2
mcol.ensembl=1df <- read.delim("../data/CYT/UMI4C/UMI4C_promoters_views.tsv", stringsAsFactors=T, header=T)
for (i in 1:length(df$bait)) {
load(file.path(out_dir, paste0("UMI4C_norm_results_", df$bait[i], ".rda")))
xlim <- c(df$start[i], df$end[i])
region <- GRanges(seqnames=paste0(seqnames(res$bait)),
ranges=IRanges(start=xlim[1],
end=xlim[2]))
### Genes ------------------
plot.genes <- plotGenes(genes=res.gr[res.gr$type!="not-expressed",],
which=region,
mcol.color=mcol.color,
mcol.name=mcol.name,
mcol.ensembl=mcol.ensembl)
g.plots <-
ggplot() +
plot.genes +
xlim(xlim) +
scale_fill_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated")) +
scale_color_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated")) +
themeXblank() +
themeYblank()
### UMI-4C ------------------
umi <-
ggplot(res$norm_trend,
aes(start, trend)) +
geom_ribbon(aes(ymin=devM, ymax=devP, group=interaction(group, sample)),
color=NA, fill="light grey") +
geom_line(aes(color=sample, group=interaction(group, sample)),
lwd=0.7) +
annotate("point", x=start(res$bait), y=df$ymax[i], pch=25, fill="black",
size=3) +
annotate("text", x=start(res$bait), y=df$ymax[i]-0.2, label=df$bait[i],
size=3) +
scale_color_manual(values=c(ctrl="#1f78b4", treat="#d95f02"),
labels=c("CYT", "CTRL"), name="") +
scale_y_continuous(name="# UMI-4C contacts",
limits=c(0, df$ymax[i]),
breaks=scales::pretty_breaks(),
expand=c(0,0)) +
xlim(xlim) +
theme(legend.position="right")
### RE ------------------
re.sel <- as.data.frame(subsetByOverlaps(resize(re, 3e3, fix="center"),
region))
ire <-
ggplot(re.sel) +
geom_rect(aes(xmin=start, xmax=end, ymin=0, ymax=1, fill=h3k27ac.log2FoldChange)) +
scale_fill_gradient2(low="dark grey",
mid="grey",
high="dark green",
name=expression("H3K27ac "*log[2]*" FC"),
breaks=scales::pretty_breaks(n=3),
midpoint=0,
guide = guide_colorbar(direction = "horizontal",
title.position="top")) +
scale_x_continuous(name=paste("Coordinates", seqnames(region), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlim,
labels=function(x) round(x/1e6, 2),
expand=c(0,0))
### Get legends -----------
gene.leg <- get_legend(g.plots)
umi.leg <- get_legend(umi)
diff.leg <- get_legend(diff$plot)
ire.leg <- get_legend(ire)
legends <- plot_grid(gene.leg, umi.leg, diff.leg, ire.leg, ncol=1)
### Grid ------------------
p <-
plot_grid(g.plots + theme(plot.margin = margin(1,0,0,0, "cm"),
legend.position = "none"),
umi + themeXblank() + theme(legend.position = "none"),
diff$plot + xlim(xlim) + themeXblank() + themeYblank() + theme(legend.position = "none"),
ire + themeYblank() + theme(legend.position = "none"),
ncol=1,
rel_heights = c(0.25, 0.45, 0.1, 0.2),
align="v")
plot <-
plot_grid(p,
legends,
ncol=2, rel_widths=c(0.7, 0.3))
print(plot)
}
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
Plot zoom-ins
TNFSF10
load(file.path(out_dir, "UMI4C_norm_results_TNFSF10.rda"))
wins <- as.character(diff$results$id[diff$results$sign=="yes"])
coord <- diff$results[diff$results$id %in% wins, c(8:9,1)]
reg <- GRanges(paste0(as.character(seqnames(res$bait)),
":", min(coord$start), "-", max(coord$end)))
tracks <- c(list.files("../data/CYT/ATAC/Visualization",
pattern=".bw",
full.names=T),
list.files("../data/CYT/H3K27ac/Visualization",
pattern=".bw",
full.names=T))
tracks <- tracks[!grepl("[[:digit:]]_", tracks) &
grepl("hi_", tracks)]## ATAC-seq -------------------
sm.at <- 20
ctrl.at <- rtracklayer::import(tracks[grepl("ATAC", tracks) & grepl("ctrl", tracks)],
which=reg)
score(ctrl.at) <- zoo::rollmean(score(ctrl.at), sm.at,
fill=c(NA, NA, NA))
cyt.at <- rtracklayer::import(tracks[grepl("ATAC", tracks) & grepl("cyt", tracks)],
which=reg)
score(cyt.at) <- zoo::rollmean(score(cyt.at), sm.at,
fill=c(NA, NA, NA))
## H3K27ac -------------------
sm.ac <- 20
ctrl.ac <- rtracklayer::import(tracks[grepl("H3K27ac", tracks) & grepl("ctrl", tracks)],
which=reg)
score(ctrl.ac) <- zoo::rollmean(score(ctrl.ac), sm.ac,
fill=c(NA, NA, NA))
cyt.ac <- rtracklayer::import(tracks[grepl("H3K27ac", tracks) & grepl("cyt", tracks)],
which=reg)
score(cyt.ac) <- zoo::rollmean(score(cyt.ac), sm.ac,
fill=c(NA, NA, NA))##--------------------
## Plot
##--------------------
xlims <- c(start(ranges(reg)),
end(ranges(reg)))
ctrl.at.p <-
ggplot(data.frame(ctrl.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,30),
expand=c(0,0),
breaks=c(0,30)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.at.p <-
ggplot(data.frame(cyt.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,30),
expand=c(0,0),
breaks=c(0,30)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
ctrl.ac.p <-
ggplot(data.frame(ctrl.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,60),
expand=c(0,0),
breaks=c(0,60)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.ac.p <-
ggplot(data.frame(cyt.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,60),
expand=c(0,0),
breaks=c(0,60)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
plot_grid(ctrl.at.p,
cyt.at.p,
ctrl.ac.p,
cyt.ac.p,
align="v",
ncol=1)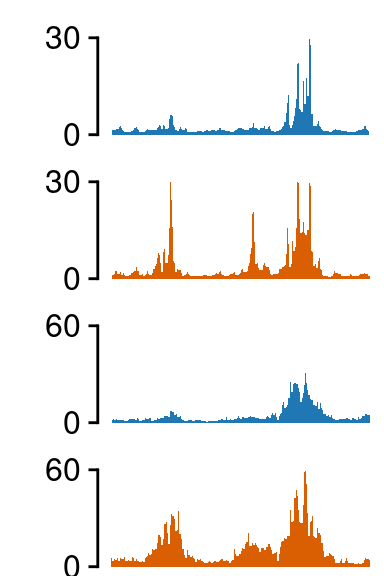
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
GBP1
Region 1
load(file.path(out_dir, "UMI4C_norm_results_GBP1.rda"))
wins <- as.character(diff$results$id[diff$results$sign=="yes"])[2]
wins <- c(wins, "window_117")
coord <- diff$results[diff$results$id %in% wins, c(8:9,1)]
reg <- GRanges(paste0(as.character(seqnames(res$bait)),
":", min(coord$start), "-", max(coord$end)))
tracks <- c(list.files("../data/CYT/ATAC/Visualization",
pattern=".bw",
full.names=T),
list.files("../data/CYT/H3K27ac/Visualization",
pattern=".bw",
full.names=T))
tracks <- tracks[!grepl("[[:digit:]]_", tracks) &
grepl("hi_", tracks)]## ATAC-seq -------------------
sm.at <- 20
ctrl.at <- rtracklayer::import(tracks[grepl("ATAC", tracks) & grepl("ctrl", tracks)],
which=reg)
score(ctrl.at) <- zoo::rollmean(score(ctrl.at), sm.at,
fill=c(NA, NA, NA))
cyt.at <- rtracklayer::import(tracks[grepl("ATAC", tracks) & grepl("cyt", tracks)],
which=reg)
score(cyt.at) <- zoo::rollmean(score(cyt.at), sm.at,
fill=c(NA, NA, NA))
## H3K27ac -------------------
sm.ac <- 20
ctrl.ac <- rtracklayer::import(tracks[grepl("H3K27ac", tracks) & grepl("ctrl", tracks)],
which=reg)
score(ctrl.ac) <- zoo::rollmean(score(ctrl.ac), sm.ac,
fill=c(NA, NA, NA))
cyt.ac <- rtracklayer::import(tracks[grepl("H3K27ac", tracks) & grepl("cyt", tracks)],
which=reg)
score(cyt.ac) <- zoo::rollmean(score(cyt.ac), sm.ac,
fill=c(NA, NA, NA))##--------------------
## Plot
##--------------------
xlims <- c(start(ranges(reg)),
end(ranges(reg)))
ctrl.at.p <-
ggplot(data.frame(ctrl.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,15),
expand=c(0,0),
breaks=c(0,15)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.at.p <-
ggplot(data.frame(cyt.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,15),
expand=c(0,0),
breaks=c(0,15)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
ctrl.ac.p <-
ggplot(data.frame(ctrl.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,10),
expand=c(0,0),
breaks=c(0,10)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.ac.p <-
ggplot(data.frame(cyt.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,10),
expand=c(0,0),
breaks=c(0,10)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
plot_grid(ctrl.at.p,
cyt.at.p,
ctrl.ac.p,
cyt.ac.p,
align="v",
ncol=1)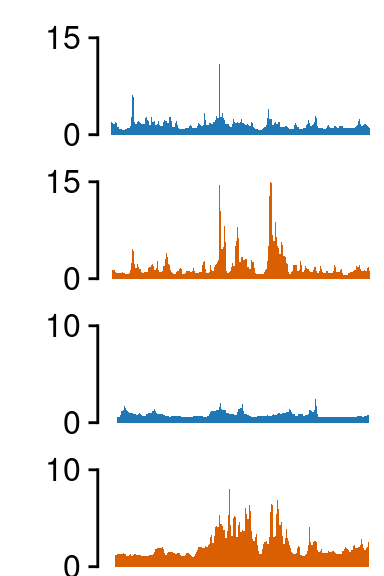
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
Region 2
load(file.path(out_dir, "UMI4C_norm_results_GBP1.rda"))
wins <- as.character(diff$results$id[diff$results$sign=="yes"])[3]
coord <- diff$results[diff$results$id %in% wins, c(8:9,1)]
reg <- GRanges(paste0(as.character(seqnames(res$bait)),
":", min(coord$start), "-", max(coord$end)))
tracks <- c(list.files("../data/CYT/ATAC/Visualization",
pattern=".bw",
full.names=T),
list.files("../data/CYT/H3K27ac/Visualization",
pattern=".bw",
full.names=T))
tracks <- tracks[!grepl("[[:digit:]]_", tracks) &
grepl("hi_", tracks)]## ATAC-seq -------------------
sm.at <- 20
ctrl.at <- rtracklayer::import(tracks[grepl("ATAC", tracks) & grepl("ctrl", tracks)],
which=reg)
score(ctrl.at) <- zoo::rollmean(score(ctrl.at), sm.at,
fill=c(NA, NA, NA))
cyt.at <- rtracklayer::import(tracks[grepl("ATAC", tracks) & grepl("cyt", tracks)],
which=reg)
score(cyt.at) <- zoo::rollmean(score(cyt.at), sm.at,
fill=c(NA, NA, NA))
## H3K27ac -------------------
sm.ac <- 20
ctrl.ac <- rtracklayer::import(tracks[grepl("H3K27ac", tracks) & grepl("ctrl", tracks)],
which=reg)
score(ctrl.ac) <- zoo::rollmean(score(ctrl.ac), sm.ac,
fill=c(NA, NA, NA))
cyt.ac <- rtracklayer::import(tracks[grepl("H3K27ac", tracks) & grepl("cyt", tracks)],
which=reg)
score(cyt.ac) <- zoo::rollmean(score(cyt.ac), sm.ac,
fill=c(NA, NA, NA))##--------------------
## Plot
##--------------------
xlims <- c(start(ranges(reg)),
end(ranges(reg)))
ctrl.at.p <-
ggplot(data.frame(ctrl.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,40),
expand=c(0,0),
breaks=c(0,40)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.at.p <-
ggplot(data.frame(cyt.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,40),
expand=c(0,0),
breaks=c(0,40)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
ctrl.ac.p <-
ggplot(data.frame(ctrl.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,60),
expand=c(0,0),
breaks=c(0,60)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.ac.p <-
ggplot(data.frame(cyt.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,60),
expand=c(0,0),
breaks=c(0,60)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
plot_grid(ctrl.at.p,
cyt.at.p,
ctrl.ac.p,
cyt.ac.p,
align="v",
ncol=1)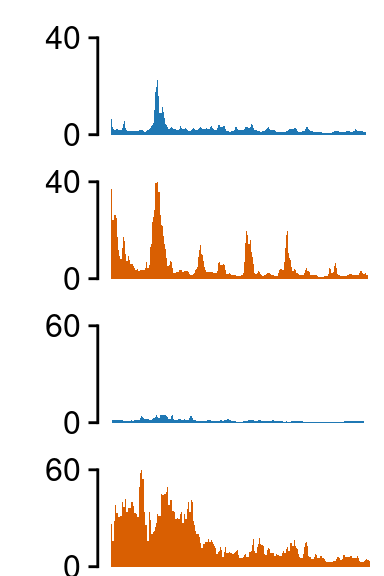
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
Region 3
load(file.path(out_dir, "UMI4C_norm_results_GBP1.rda"))
wins <- as.character(diff$results$id[diff$results$sign=="yes"])[4]
coord <- diff$results[diff$results$id %in% wins, c(8:9,1)]
reg <- GRanges(paste0(as.character(seqnames(res$bait)),
":", min(coord$start), "-", max(coord$end)))
tracks <- c(list.files("../data/CYT/ATAC/Visualization",
pattern=".bw",
full.names=T),
list.files("../data/CYT/H3K27ac/Visualization",
pattern=".bw",
full.names=T))
tracks <- tracks[!grepl("[[:digit:]]_", tracks) &
grepl("hi_", tracks)]## ATAC-seq -------------------
sm.at <- 20
ctrl.at <- rtracklayer::import(tracks[grepl("ATAC", tracks) & grepl("ctrl", tracks)],
which=reg)
score(ctrl.at) <- zoo::rollmean(score(ctrl.at), sm.at,
fill=c(NA, NA, NA))
cyt.at <- rtracklayer::import(tracks[grepl("ATAC", tracks) & grepl("cyt", tracks)],
which=reg)
score(cyt.at) <- zoo::rollmean(score(cyt.at), sm.at,
fill=c(NA, NA, NA))
## H3K27ac -------------------
sm.ac <- 20
ctrl.ac <- rtracklayer::import(tracks[grepl("H3K27ac", tracks) & grepl("ctrl", tracks)],
which=reg)
score(ctrl.ac) <- zoo::rollmean(score(ctrl.ac), sm.ac,
fill=c(NA, NA, NA))
cyt.ac <- rtracklayer::import(tracks[grepl("H3K27ac", tracks) & grepl("cyt", tracks)],
which=reg)
score(cyt.ac) <- zoo::rollmean(score(cyt.ac), sm.ac,
fill=c(NA, NA, NA))##--------------------
## Plot
##--------------------
xlims <- c(start(ranges(reg)),
end(ranges(reg)))
ctrl.at.p <-
ggplot(data.frame(ctrl.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,60),
expand=c(0,0),
breaks=c(0,60)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.at.p <-
ggplot(data.frame(cyt.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,60),
expand=c(0,0),
breaks=c(0,60)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
ctrl.ac.p <-
ggplot(data.frame(ctrl.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,50),
expand=c(0,0),
breaks=c(0,50)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.ac.p <-
ggplot(data.frame(cyt.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,50),
expand=c(0,0),
breaks=c(0,50)) +
xlim(xlims) +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
plot_grid(ctrl.at.p,
cyt.at.p,
ctrl.ac.p,
cyt.ac.p,
align="v",
ncol=1)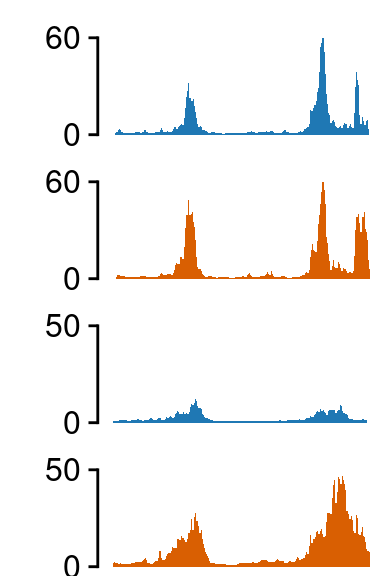
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
Distribution of UMI-4C contact changes at promoters of up-regulated genes
files <- list.files(out_dir,
pattern="test",
full.names=T)
test.all <- data.frame()
for (i in files) {
load(i)
test <- test[,c(4,21,25)]
name <- pipelineNGS::getNameFromPath(i, prefix="UMI4C_test_promoters_",
suffix=".rda")
test$bait <- name
test.all <- rbind(test.all, test)
}
test.all <- test.all[!grepl("Other", test.all$subgroup2),]ggplot(test.all,
aes(subgroup2, log2_foldChange)) +
geom_hline(yintercept=0, lty=2, color="grey") +
geom_boxplot(aes(color=subgroup2), notch=T, lwd=1) +
scale_color_manual(values=pals$re) +
scale_x_discrete(name="RE type",
labels=function(x) paste0(x, "s")) +
ylab(expression(Log[2]~FC~UMI4C~contacts)) +
theme(legend.position="none",
axis.text.x=element_text(angle=30, hjust=1))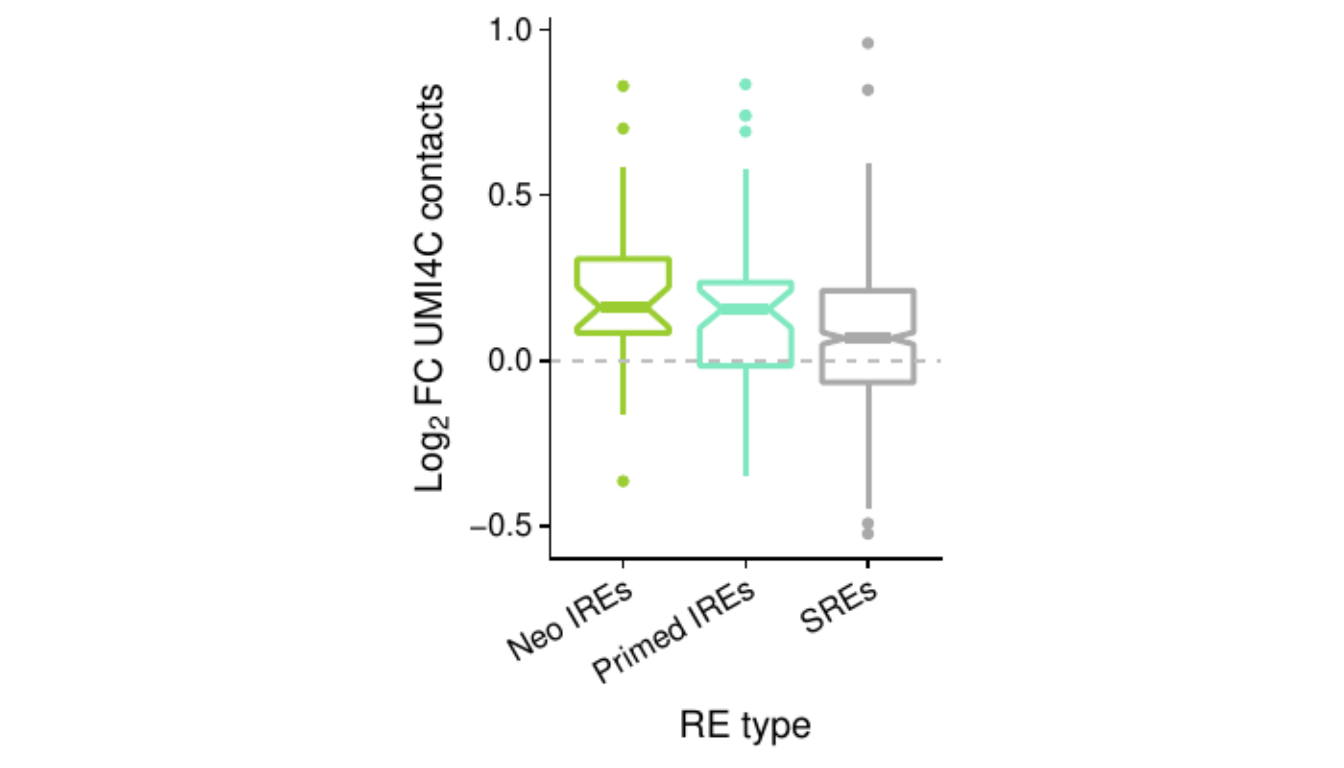
| Version | Author | Date |
|---|---|---|
| 90643f9 | mireia-bioinfo | 2020-08-31 |
sessionInfo()R version 4.0.2 (2020-06-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.1 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=es_ES.UTF-8 LC_COLLATE=C
[5] LC_MONETARY=es_ES.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=es_ES.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=es_ES.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] umi4cCatcheR_0.0.0.9000 RColorBrewer_1.1-2 GenomicRanges_1.41.5
[4] GenomeInfoDb_1.25.8 IRanges_2.23.10 S4Vectors_0.27.12
[7] BiocGenerics_0.35.4 cowplot_1.0.0 ggplot2_3.3.2
[10] dplyr_1.0.1 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] bitops_1.0-6 matrixStats_0.56.0
[3] fs_1.5.0 usethis_1.6.1
[5] devtools_2.3.1 rprojroot_1.3-2
[7] tools_4.0.2 backports_1.1.8
[9] R6_2.4.1 colorspace_1.4-1
[11] withr_2.2.0 tidyselect_1.1.0
[13] gridExtra_2.3 prettyunits_1.1.1
[15] processx_3.4.3 compiler_4.0.2
[17] git2r_0.27.1 cli_2.0.2
[19] Biobase_2.49.0 desc_1.2.0
[21] DelayedArray_0.15.7 rtracklayer_1.49.4
[23] labeling_0.3 bookdown_0.20
[25] scales_1.1.1 callr_3.4.3
[27] askpass_1.1 stringr_1.4.0
[29] digest_0.6.25 Rsamtools_2.5.3
[31] rmarkdown_2.3 XVector_0.29.3
[33] pkgconfig_2.0.3 htmltools_0.5.0
[35] sessioninfo_1.1.1 rlang_0.4.7
[37] rstudioapi_0.11 generics_0.0.2
[39] farver_2.0.3 zoo_1.8-8
[41] BiocParallel_1.23.2 RCurl_1.98-1.2
[43] magrittr_1.5 GenomeInfoDbData_1.2.3
[45] Matrix_1.2-18 Rcpp_1.0.5
[47] munsell_0.5.0 fansi_0.4.1
[49] lifecycle_0.2.0 stringi_1.4.6
[51] whisker_0.4 yaml_2.2.1
[53] SummarizedExperiment_1.19.6 zlibbioc_1.35.0
[55] pkgbuild_1.1.0 grid_4.0.2
[57] promises_1.1.1 crayon_1.3.4
[59] lattice_0.20-41 Biostrings_2.57.2
[61] magick_2.4.0 knitr_1.29
[63] ps_1.3.3 pillar_1.4.6
[65] pkgload_1.1.0 XML_3.99-0.5
[67] glue_1.4.1 evaluate_0.14
[69] pdftools_2.3.1 qpdf_1.1
[71] remotes_2.2.0 vctrs_0.3.2
[73] httpuv_1.5.4 testthat_2.3.2
[75] gtable_0.3.0 purrr_0.3.4
[77] assertthat_0.2.1 xfun_0.16
[79] later_1.1.0.1 tibble_3.0.3
[81] GenomicAlignments_1.25.3 memoise_1.1.0
[83] ellipsis_0.3.1