#5 IREs and T1D risk
Islet induced regulotory elements are implicated in T1D genetic susceptibility
Mireia Ramos-Rodríguez
Details
- Original publication:
Ramos-Rodríguez, M., Raurell-Vila, H., Colli, M.L. et al. The impact of proinflammatory cytokines on the β-cell regulatory landscape provides insights into the genetics of type 1 diabetes. Nat Genet. 51, 1588–1595 (2019) https://doi.org/10.1038/s41588-019-0524-6
Contents: Analyses and figures contained in this document correspond to the following figures/sections of the original publication:
- Results: “Islet cytokine enhancers are implicated in T1D genetic susceptibility”.
- Figure 4: “Cytokine-induced islet regulatory elements map to T1D-associated regions and guide the identification of functional risk variants”. Panels a, b, d, e, f and h.
- Extended Data Figure 6: “Cytokine-induced islet regulatory elements are enriched in T1D associated variants”. Panels a to f.
Methods
- Data was downloaded from the GWAS catalog by selecting trait Type i diabetes mellitus with child traits. Date: 2019-04-18
- Only SNPs with
DISEASE.TRAIT=="Type 1 diabetes"were kept. - Proxies were obtained using
proxysnpsare package, which obtains proxies using 1,000 Genomes phase 3, EUR population andwindow_size=2.5e5. Only proxies with LD >= 0.5 were kept. - Locus were determined creating a window from the most upstream to the most downstream SNP. Overlapping loci were merged.
Rscript code/CYT_create_LD0.8.R
Rscript code/CYT_create_T1D_loci_public.R
Rscript code/CYT_create_T2D_loci_public.R
Rscript code/CYT_rm_common_T1D_T2D.Rload("../data/CYT/T1D_GWAS/T1D_LD0.8_risk_loci_granges.rda")
df <- data.frame(loci)
df$seqnames <- factor(df$seqnames, levels=rev(paste0("chr", c(1:22, "X", "Y"))))
width <-
ggplot(df,
aes(width)) +
geom_density() +
scale_x_continuous(labels=scales::comma,
name="Locus width (bp)")
counts <-
ggplot(df,
aes(seqnames)) +
geom_dotplot(aes(fill=seqnames)) +
coord_flip() +
xlab("Chromosome") +
ylab("Proportion of loci") +
theme(legend.position="none")
plot_grid(counts, width, ncol=2, rel_widths=c(.35, .65))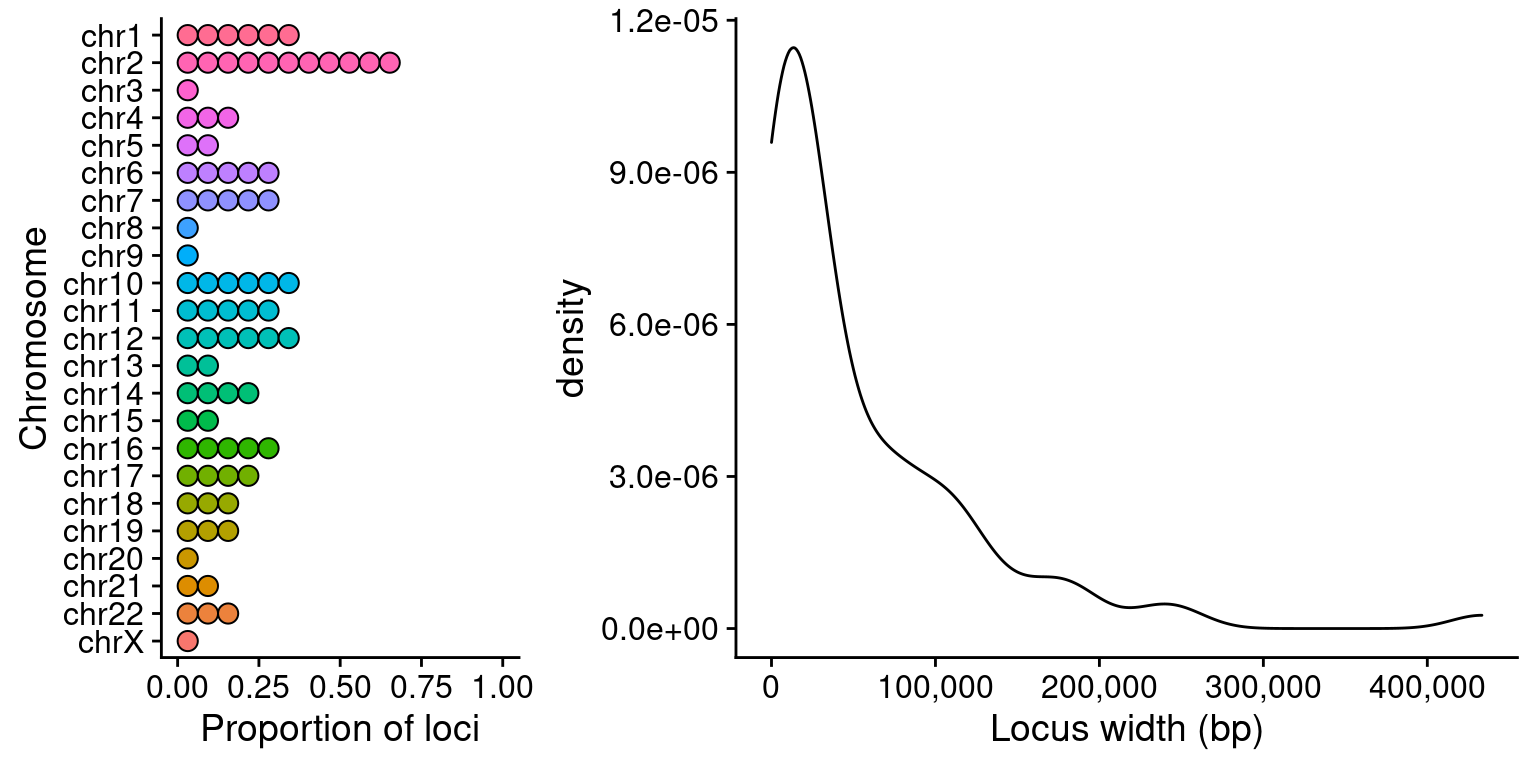
| Version | Author | Date |
|---|---|---|
| 986b505 | mireia-bioinfo | 2020-09-01 |
Variant Set Enrichment Analysis (VSE)
T1D GWAS data minus loci common with T2D
re <- list.files("../data/CYT/bedfiles",
pattern="*.bed",
full.names=T)
re <- re[!grepl("distal", re)]
re <- re[grepl("padj0.05", re) &
grepl("hi", re)]
samples <- data.frame(X=1:length(re),
SampleID=pipelineNGS::getNameFromPath(re, suffix=".bed"),
Peaks=re)
samples$SampleID <- gsub("_fc1_padj0.05", "", samples$SampleID)
load("../data/CYT/T1D_GWAS/T1Drm_VSE_MRVS_500.rda")
load("../data/CYT/T1D_GWAS/T1Drm_VSE_AVS.rda")
seqlevelsStyle(t1d.avs) <- "Ensembl"
t1d.vse <- variantSetEnrichment.mod(t1d.avs, t1d.mrvs.500, samples)
save(t1d.vse, file=file.path(out_dir, "T1Drm_hi_vse_result.rda"))load(file.path(out_dir, "T1Drm_hi_vse_result.rda"))
t1d.vse.res <- VSESummary.mod(t1d.vse,
method="bonferroni")
knitr::kable(t1d.vse.res)| region | avs | enrichment | p.value | p.adjusted | |
|---|---|---|---|---|---|
| IREs_hi | IREs_hi | 13 | 4.617614 | 0.0000015 | 0.0000030 |
| SREs_hi | SREs_hi | 36 | 1.139202 | 0.1290808 | 0.2581615 |
VSEplot.mod(t1d.vse,
method="bonferroni",
pch = 20,
cex = 1,
padj = 0.05,
main = "T1D in HI")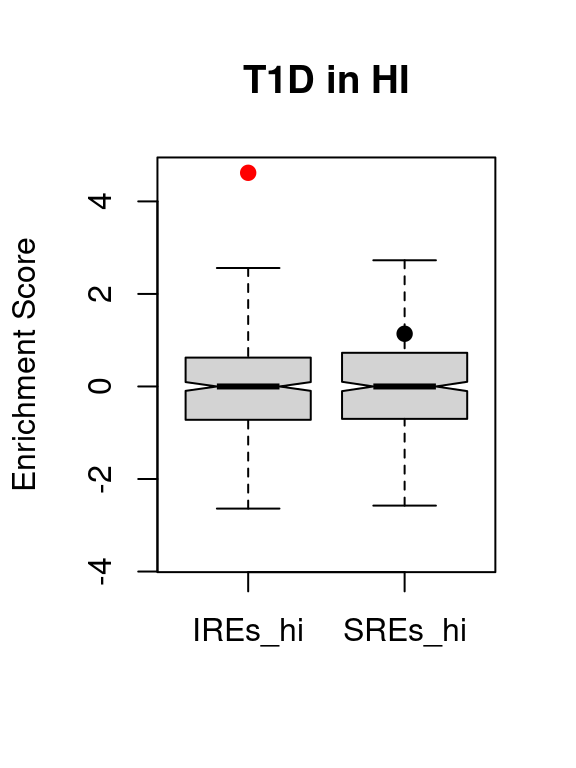
| Version | Author | Date |
|---|---|---|
| 986b505 | mireia-bioinfo | 2020-09-01 |
re <- list.files("../data/CYT/bedfiles",
pattern="*.bed",
full.names=T)
re <- re[!grepl("distal", re) & !grepl("k270.8", re)]
re <- re[grepl("padj0.05", re) &
grepl("endoc", re)]
samples <- data.frame(X=1:length(re),
SampleID=pipelineNGS::getNameFromPath(re, suffix=".bed"),
Peaks=re)
samples$SampleID <- gsub("_fc1_padj0.05", "", samples$SampleID)
load("../data/CYT/T1D_GWAS/T1Drm_VSE_MRVS_500.rda")
load("../data/CYT/T1D_GWAS/T1Drm_VSE_AVS.rda")
seqlevelsStyle(t1d.avs) <- "Ensembl"
t1d.vse <- variantSetEnrichment.mod(t1d.avs, t1d.mrvs.500, samples)
save(t1d.vse, file=file.path(out_dir, "T1Drm_endoc_vse_result.rda"))load(file.path(out_dir, "T1Drm_endoc_vse_result.rda"))
t1d.vse.res <- VSESummary.mod(t1d.vse,
method="bonferroni")
knitr::kable(t1d.vse.res)| region | avs | enrichment | p.value | p.adjusted | |
|---|---|---|---|---|---|
| IREs_endoc | IREs_endoc | 8 | 3.140784 | 0.0007120 | 0.0014240 |
| SREs_endoc | SREs_endoc | 33 | 1.810616 | 0.0342564 | 0.0685129 |
VSEplot.mod(t1d.vse,
method="bonferroni",
pch = 20,
cex = 1,
padj = 0.05,
main = "T1D in EndoC")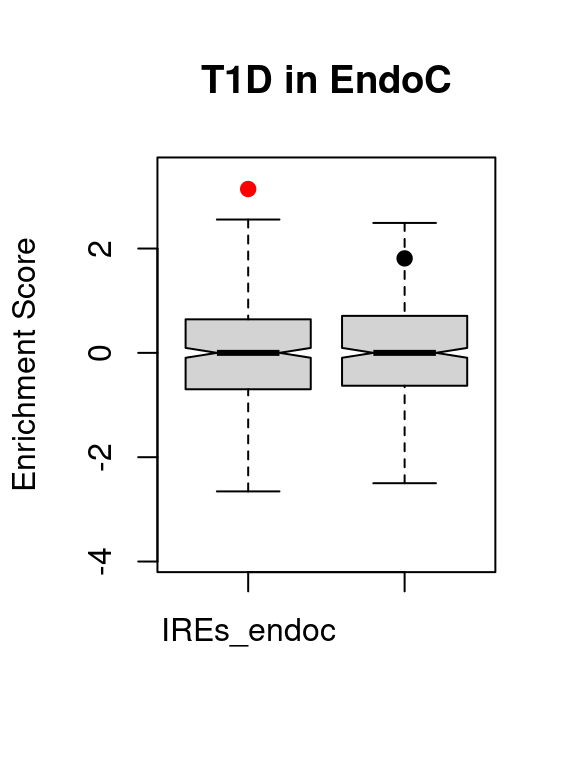
| Version | Author | Date |
|---|---|---|
| 986b505 | mireia-bioinfo | 2020-09-01 |
T2D GWAS data minus loci common with T1D
re <- list.files("../data/CYT/bedfiles",
pattern="*.bed",
full.names=T)
re <- re[!grepl("distal", re)]
re <- re[grepl("padj0.05", re) &
grepl("hi", re)]
samples <- data.frame(X=1:length(re),
SampleID=pipelineNGS::getNameFromPath(re, suffix=".bed"),
Peaks=re)
samples$SampleID <- gsub("_fc1_padj0.05", "", samples$SampleID)
load("../data/CYT/T1D_GWAS/T2Drm_VSE_MRVS_500.rda")
load("../data/CYT/T1D_GWAS/T2Drm_VSE_AVS.rda")
seqlevelsStyle(t2d.avs) <- "Ensembl"
t2d.vse <- variantSetEnrichment.mod(t2d.avs, t2d.mrvs.500, samples)
save(t2d.vse, file=file.path(out_dir, "T2Drm_hi_vse_result.rda"))load(file.path(out_dir, "T2Drm_hi_vse_result.rda"))
t2d.vse.res <- VSESummary.mod(t2d.vse,
method="bonferroni")
knitr::kable(t2d.vse.res)| region | avs | enrichment | p.value | p.adjusted | |
|---|---|---|---|---|---|
| IREs_hi | IREs_hi | 18 | -0.3737613 | 0.6490075 | 1 |
| SREs_hi | SREs_hi | 216 | 5.4742895 | 0.0000000 | 0 |
VSEplot.mod(t2d.vse,
method="bonferroni",
pch = 20,
cex = 1,
padj = 0.05,
main = "T2D in HI")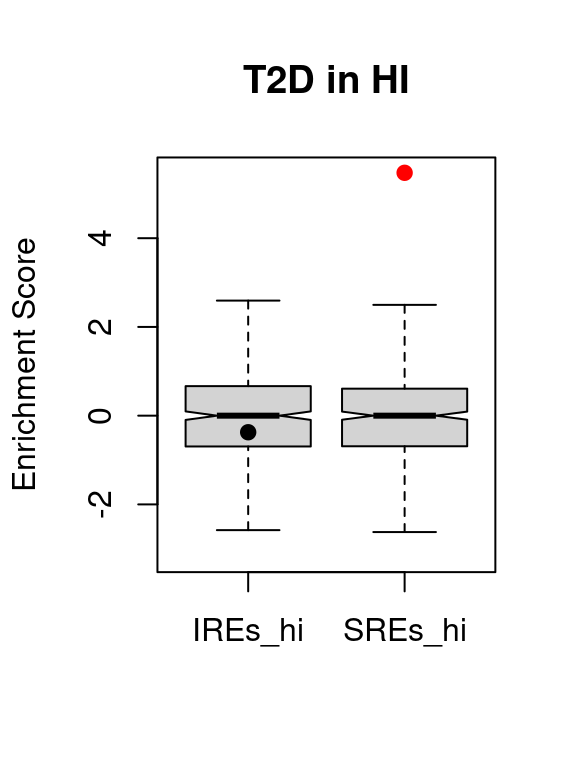
| Version | Author | Date |
|---|---|---|
| 986b505 | mireia-bioinfo | 2020-09-01 |
re <- list.files("../data/CYT/bedfiles",
pattern="*.bed",
full.names=T)
re <- re[!grepl("distal", re) & !grepl("k270.8", re)]
re <- re[grepl("padj0.05", re) &
grepl("endoc", re)]
samples <- data.frame(X=1:length(re),
SampleID=pipelineNGS::getNameFromPath(re, suffix=".bed"),
Peaks=re)
samples$SampleID <- gsub("_fc1_padj0.05", "", samples$SampleID)
load("../data/CYT/T1D_GWAS/T2Drm_VSE_MRVS_500.rda")
load("../data/CYT/T1D_GWAS/T2Drm_VSE_AVS.rda")
seqlevelsStyle(t2d.avs) <- "Ensembl"
t2d.vse <- variantSetEnrichment.mod(t2d.avs, t2d.mrvs.500, samples)
save(t2d.vse, file=file.path(out_dir, "T2Drm_endoc_vse_result.rda"))load(file.path(out_dir, "T2Drm_endoc_vse_result.rda"))
t2d.vse.res <- VSESummary.mod(t2d.vse,
method="bonferroni")
knitr::kable(t2d.vse.res)| region | avs | enrichment | p.value | p.adjusted | |
|---|---|---|---|---|---|
| IREs_endoc | IREs_endoc | 19 | 0.6608974 | 0.2572423 | 0.5144847 |
| SREs_endoc | SREs_endoc | 174 | 4.5283477 | 0.0000029 | 0.0000059 |
VSEplot.mod(t2d.vse,
method="bonferroni",
pch = 20,
cex = 1,
padj = 0.05,
main = "T2D in EndoC")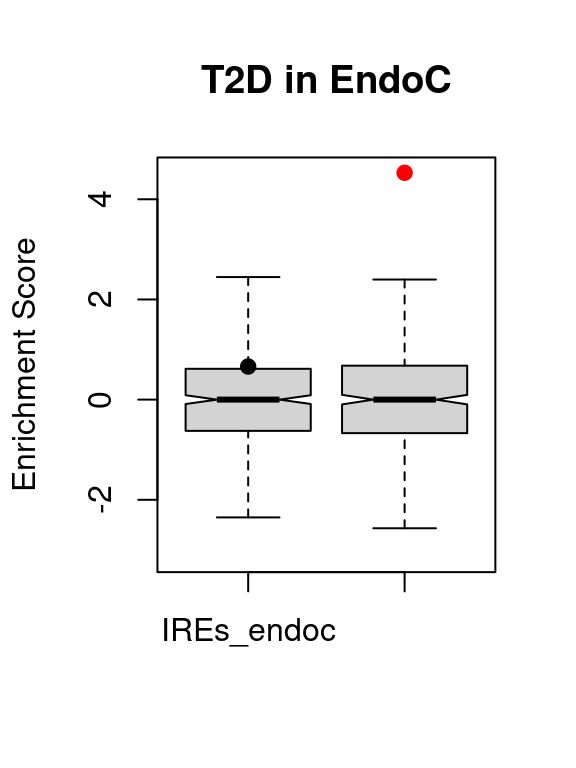
| Version | Author | Date |
|---|---|---|
| 986b505 | mireia-bioinfo | 2020-09-01 |
Views of example T1D loci with variants overlapping IREs
IL7R locus
## SNPs ----------------
rsids <- c("rs11954020", "rs6897932")
load("../data/CYT/T1D_GWAS/T1Drm_LD0.8_VSE_AVS.rda")
load("../data/CYT/T1D_GWAS/T1Drm_risk_proxies_granges.rda")
t1d.dat <- data.frame(mcols(proxies))[,c(1,5)]
t1d.dat <- t1d.dat[t1d.dat$R.squared>=0.8,]
snp <- unlist(t1d.avs)
snp <- data.frame(snp[snp$idTag %in% rsids,])
snp$type <- "proxy"
snp$type[snp$idTag==snp$idLd] <- "leading"
snp$lab <- ""
snp$lab[snp$idLd %in% c(rsids, "rs34143578", "rs6890853", "rs10214273")] <- snp$idLd[snp$idLd %in% c(rsids, "rs34143578", "rs6890853", "rs10214273")]
snp <- dplyr::left_join(snp,
t1d.dat,
by=c("idLd"="ID"))
snp$type <- factor(snp$type, levels=c("proxy", "leading"))
snp <- snp[order(snp$type),]
win <- GRanges(paste0(unique(snp$seqnames), ":", min(snp$start), "-", max(snp$start)))
xlims <- c(min(snp$start), max(snp$start))
## RE --------------------
load("../data/CYT/REs/REs_hi_fc1_padj0.05_granges_subgroup.rda")
re.sel <- subsetByOverlaps(re, win)
re.sel <- resize(re.sel, width=1e3, fix="center")
re.sel <- data.frame(re.sel)
## Genes -------------------
load("../data/CYT/RNA/diffAnalysis/RNA_hi_GRangesBatch.rda")
res.gr$type[res.gr$baseMean<=1] <- "not-expressed"
res.gr <- res.gr[res.gr$gene_biotype=="protein_coding",]
col.df <- data.frame("type"=c(names(pals$differential), "not-expressed"),
"color"=c(pals$differential, "black"),
stringsAsFactors = FALSE)
col.df$color[grep("grey", col.df$color)] <- "grey39"
mcols(res.gr) <- dplyr::left_join(data.frame(mcols(res.gr)[,c(1:2,10)]),
col.df)
colnames(mcols(res.gr))[1] <- "ensembl_gene_id"
mcol.color=4
mcol.name=2
mcol.ensembl=1## Plot snps ------------------
snp.plot <-
ggplot(snp,
aes(start, R.squared)) +
geom_vline(xintercept=snp$start[snp$type=="proxy" & snp$lab!=""],
lty=2, color="grey") +
geom_point(aes(shape=type, size=R.squared, fill=idTag)) +
scale_shape_manual(values=c(21, 23), name="Type of variant") +
scale_size_continuous(guide=F) +
scale_fill_discrete(name="Leading variant") +
ggrepel::geom_text_repel(aes(label=lab)) +
scale_y_continuous(name=expression(R^2),
limits=c(0.8,1),
expand=c(0,0)) +
xlim(xlims) +
themeXblank()
## Plot REs ---------------------
re.plot <-
ggplot(re.sel) +
geom_rect(aes(xmin=start, xmax=end,
ymin=0, ymax=1, fill=h3k27ac.log2FoldChange)) +
scale_fill_gradient2(low="dark grey",
mid="grey",
high="dark green",
name=expression("H3K27ac "*log[2]*" FC"),
breaks=scales::pretty_breaks(n=3),
midpoint=0,
guide = guide_colorbar(direction = "horizontal",
title.position="top")) +
xlim(xlims) +
themeYblank() +
themeXblank()
## Plot genes ------------------
plot.genes <- plotGenes(genes=res.gr[res.gr$type!="not-expressed",],
which=win,
mcol.color=mcol.color,
mcol.name=mcol.name,
mcol.ensembl=mcol.ensembl)
g.plots <-
ggplot() +
plot.genes +
scale_x_continuous(name=paste("Coordinates", seqnames(win), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlims,
labels=function(x) round(x/1e6, 2),
expand=c(0,0)) +
scale_fill_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated")) +
scale_color_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated")) +
themeYblank()
## Create legend --------------
snps.leg <- get_legend(snp.plot)
re.leg <- get_legend(re.plot)
gene.leg <- get_legend(g.plots)
legends <- plot_grid(snps.leg, re.leg, gene.leg, ncol=1)
## Plot all -------------------
main <-
plot_grid(snp.plot + theme(legend.position="none",
plot.margin=margin(1,0,0,0, "cm")),
re.plot + theme(legend.position="none"),
g.plots + theme(legend.position="none"),
ncol=1, align="v", rel_heights=c(0.65, 0.1, 0.25))
plot_grid(main, legends, rel_widths=c(0.7, 0.3))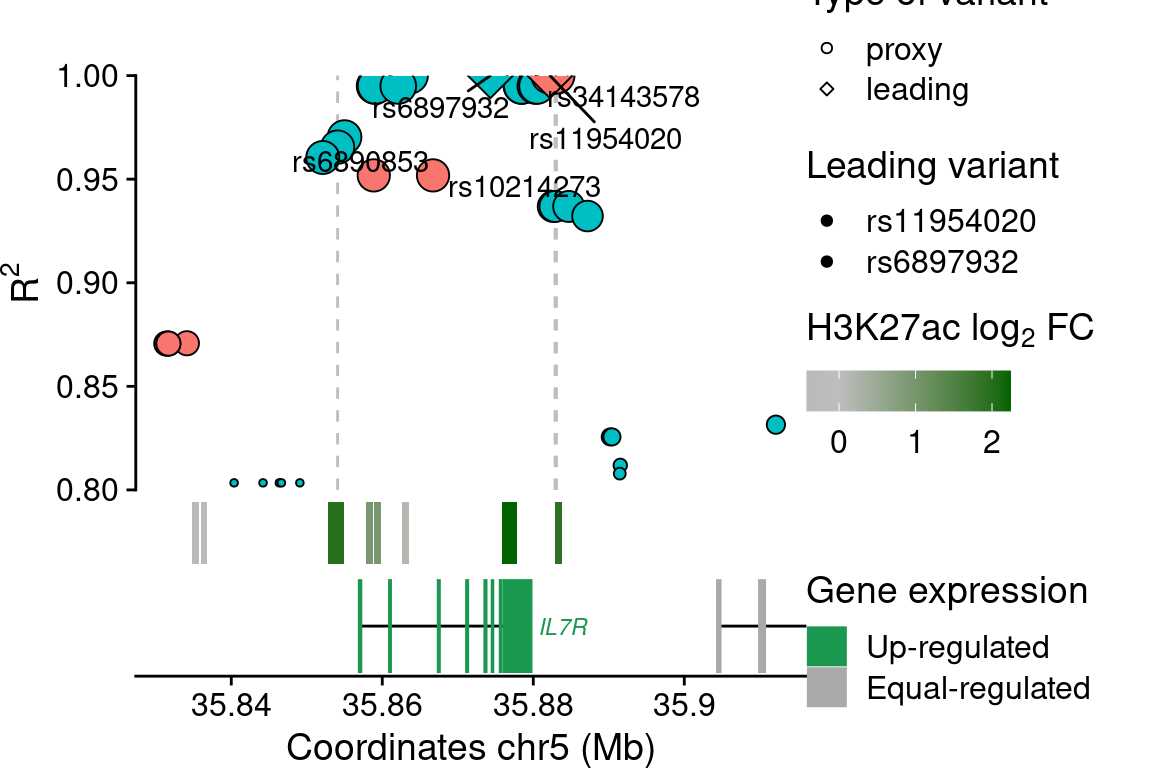
| Version | Author | Date |
|---|---|---|
| 986b505 | mireia-bioinfo | 2020-09-01 |
FGF2
## SNPs ----------------
rsids <- c("rs3024505")
load("../data/CYT/T1D_GWAS/T1Drm_LD0.8_VSE_AVS.rda")
load("../data/CYT/T1D_GWAS/T1Drm_risk_proxies_granges.rda")
t1d.dat <- data.frame(mcols(proxies))[,c(1,5)]
t1d.dat <- t1d.dat[t1d.dat$R.squared>=0.8,]
snp <- unlist(t1d.avs)
snp <- data.frame(snp[snp$idTag %in% rsids,])
snp$type <- "proxy"
snp$type[snp$idTag==snp$idLd] <- "leading"
snp$lab <- ""
snp$lab[snp$idLd %in% c(rsids, "rs3024493")] <- snp$idLd[snp$idLd %in% c(rsids, "rs3024493")]
snp <- dplyr::left_join(snp,
t1d.dat,
by=c("idLd"="ID"))
snp$type <- factor(snp$type, levels=c("proxy", "leading"))
snp <- snp[order(snp$type),]
win <- GRanges(paste0(unique(snp$seqnames), ":", min(snp$start), "-", max(snp$start)))
win <- resize(win, width=81e3, fix="start")
xlims <- c(start(win), end(win))
## RE --------------------
load("../data/CYT/REs/REs_hi_fc1_padj0.05_granges_subgroup.rda")
re.sel <- subsetByOverlaps(re, win)
re.sel <- resize(re.sel, width=1e3, fix="center")
re.sel <- data.frame(re.sel)
## Genes -------------------
load("../data/CYT/RNA/diffAnalysis/RNA_hi_GRangesBatch.rda")
res.gr$type[res.gr$baseMean<=1] <- "not-expressed"
res.gr <- res.gr[res.gr$gene_biotype=="protein_coding",]
col.df <- data.frame("type"=c(names(pals$differential), "not-expressed"),
"color"=c(pals$differential, "black"),
stringsAsFactors = FALSE)
col.df$color[grep("grey", col.df$color)] <- "grey39"
mcols(res.gr) <- dplyr::left_join(data.frame(mcols(res.gr)[,c(1:2,10)]),
col.df)
colnames(mcols(res.gr))[1] <- "ensembl_gene_id"
mcol.color=4
mcol.name=2
mcol.ensembl=1## Plot snps ------------------
snp.plot <-
ggplot(snp,
aes(start, R.squared)) +
geom_vline(xintercept=snp$start[snp$type=="proxy" & snp$lab!=""],
lty=2, color="grey") +
geom_point(aes(shape=type, size=R.squared, fill=idTag)) +
scale_shape_manual(values=c(21, 23), name="Type of variant") +
scale_size_continuous(guide=F) +
scale_fill_discrete(name="Leading variant") +
ggrepel::geom_text_repel(aes(label=lab)) +
scale_y_continuous(name=expression(R^2),
limits=c(0.8,1),
expand=c(0,0)) +
xlim(xlims) +
themeXblank()
## Plot REs ---------------------
re.plot <-
ggplot(re.sel) +
geom_rect(aes(xmin=start, xmax=end,
ymin=0, ymax=1, fill=h3k27ac.log2FoldChange)) +
scale_fill_gradient2(low="dark grey",
mid="grey",
high="dark green",
name=expression("H3K27ac "*log[2]*" FC"),
breaks=scales::pretty_breaks(n=3),
midpoint=0,
guide = guide_colorbar(direction = "horizontal",
title.position="top")) +
xlim(xlims) +
themeYblank() +
themeXblank()
## Plot genes ------------------
plot.genes <- plotGenes(genes=res.gr[res.gr$type!="not-expressed",],
which=win,
mcol.color=mcol.color,
mcol.name=mcol.name,
mcol.ensembl=mcol.ensembl)
g.plots <-
ggplot() +
plot.genes +
scale_x_continuous(name=paste("Coordinates", seqnames(win), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlims,
labels=function(x) round(x/1e6, 2),
expand=c(0,0)) +
scale_fill_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated")) +
scale_color_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated")) +
themeYblank()
## Create legend --------------
snps.leg <- get_legend(snp.plot)
re.leg <- get_legend(re.plot)
gene.leg <- get_legend(g.plots)
legends <- plot_grid(snps.leg, re.leg, gene.leg, ncol=1)
## Plot all -------------------
main <-
plot_grid(snp.plot + theme(legend.position="none",
plot.margin=margin(1,0,0,0, "cm")),
re.plot + theme(legend.position="none"),
g.plots + theme(legend.position="none"),
ncol=1, align="v", rel_heights=c(0.65, 0.1, 0.25))
plot_grid(main, legends, rel_widths=c(0.7, 0.3))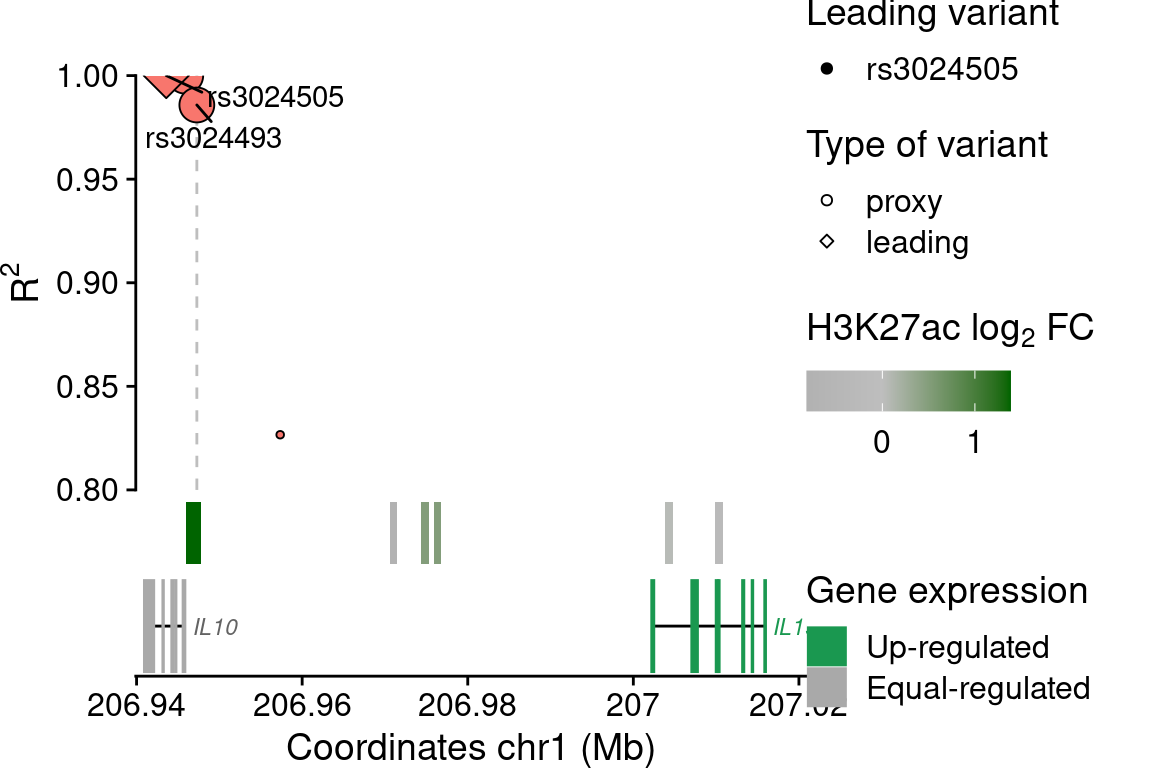
| Version | Author | Date |
|---|---|---|
| 986b505 | mireia-bioinfo | 2020-09-01 |
PTPN2
## SNPs ----------------
rsids <- c("rs12971201")
load("../data/CYT/T1D_GWAS/T1Drm_LD0.8_VSE_AVS.rda")
load("../data/CYT/T1D_GWAS/T1Drm_risk_proxies_granges.rda")
t1d.dat <- data.frame(mcols(proxies))[,c(1,5)]
t1d.dat <- t1d.dat[t1d.dat$R.squared>=0.8,]
snp <- unlist(t1d.avs)
snp <- data.frame(snp[snp$idTag %in% rsids,])
snp$type <- "proxy"
snp$type[snp$idTag==snp$idLd] <- "leading"
snp$lab <- ""
snp$lab[snp$idLd %in% c(rsids, "rs2847281")] <- snp$idLd[snp$idLd %in% c(rsids, "rs2847281")]
snp <- dplyr::left_join(snp,
t1d.dat,
by=c("idLd"="ID"))
snp$type <- factor(snp$type, levels=c("proxy", "leading"))
snp <- snp[order(snp$type),]
win <- GRanges(paste0(unique(snp$seqnames), ":", min(snp$start), "-", max(snp$start)))
win <- resize(win, width=150e3, fix="center")
xlims <- c(start(win), end(win))
## RE --------------------
load("../data/CYT/REs/REs_hi_fc1_padj0.05_granges_subgroup.rda")
re.sel <- subsetByOverlaps(re, win)
re.sel <- resize(re.sel, width=1e3, fix="center")
re.sel <- data.frame(re.sel)
## Genes -------------------
load("../data/CYT/RNA/diffAnalysis/RNA_hi_GRangesBatch.rda")
res.gr$type[res.gr$baseMean<=1] <- "not-expressed"
res.gr <- res.gr[res.gr$gene_biotype=="protein_coding",]
col.df <- data.frame("type"=c(names(pals$differential), "not-expressed"),
"color"=c(pals$differential, "black"),
stringsAsFactors = FALSE)
col.df$color[grep("grey", col.df$color)] <- "grey39"
mcols(res.gr) <- dplyr::left_join(data.frame(mcols(res.gr)[,c(1:2,10)]),
col.df)
colnames(mcols(res.gr))[1] <- "ensembl_gene_id"
mcol.color=4
mcol.name=2
mcol.ensembl=1## Plot snps ------------------
snp.plot <-
ggplot(snp,
aes(start, R.squared)) +
geom_vline(xintercept=snp$start[snp$type=="proxy" & snp$lab!=""],
lty=2, color="grey") +
geom_point(aes(shape=type, size=R.squared, fill=idTag)) +
scale_shape_manual(values=c(21, 23), name="Type of variant") +
scale_size_continuous(guide=F) +
scale_fill_discrete(name="Leading variant") +
ggrepel::geom_text_repel(aes(label=lab)) +
scale_y_continuous(name=expression(R^2),
limits=c(0.8,1),
expand=c(0,0)) +
xlim(xlims) +
themeXblank()
## Plot REs ---------------------
re.plot <-
ggplot(re.sel) +
geom_rect(aes(xmin=start, xmax=end,
ymin=0, ymax=1, fill=h3k27ac.log2FoldChange)) +
scale_fill_gradient2(low="dark grey",
mid="grey",
high="dark green",
name=expression("H3K27ac "*log[2]*" FC"),
breaks=scales::pretty_breaks(n=3),
midpoint=0,
guide = guide_colorbar(direction = "horizontal",
title.position="top")) +
xlim(xlims) +
themeYblank() +
themeXblank()
## Plot genes ------------------
plot.genes <- plotGenes(genes=res.gr[res.gr$type!="not-expressed",],
which=win,
mcol.color=mcol.color,
mcol.name=mcol.name,
mcol.ensembl=mcol.ensembl)
g.plots <-
ggplot() +
plot.genes +
scale_x_continuous(name=paste("Coordinates", seqnames(win), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlims,
labels=function(x) round(x/1e6, 2),
expand=c(0,0)) +
scale_fill_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated")) +
scale_color_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated")) +
themeYblank()
## Create legend --------------
snps.leg <- get_legend(snp.plot)
re.leg <- get_legend(re.plot)
gene.leg <- get_legend(g.plots)
legends <- plot_grid(snps.leg, re.leg, gene.leg, ncol=1)
## Plot all -------------------
main <-
plot_grid(snp.plot + theme(legend.position="none",
plot.margin=margin(1,0,0,0, "cm")),
re.plot + theme(legend.position="none"),
g.plots + theme(legend.position="none"),
ncol=1, align="v", rel_heights=c(0.65, 0.1, 0.25))
plot_grid(main, legends, rel_widths=c(0.7, 0.3))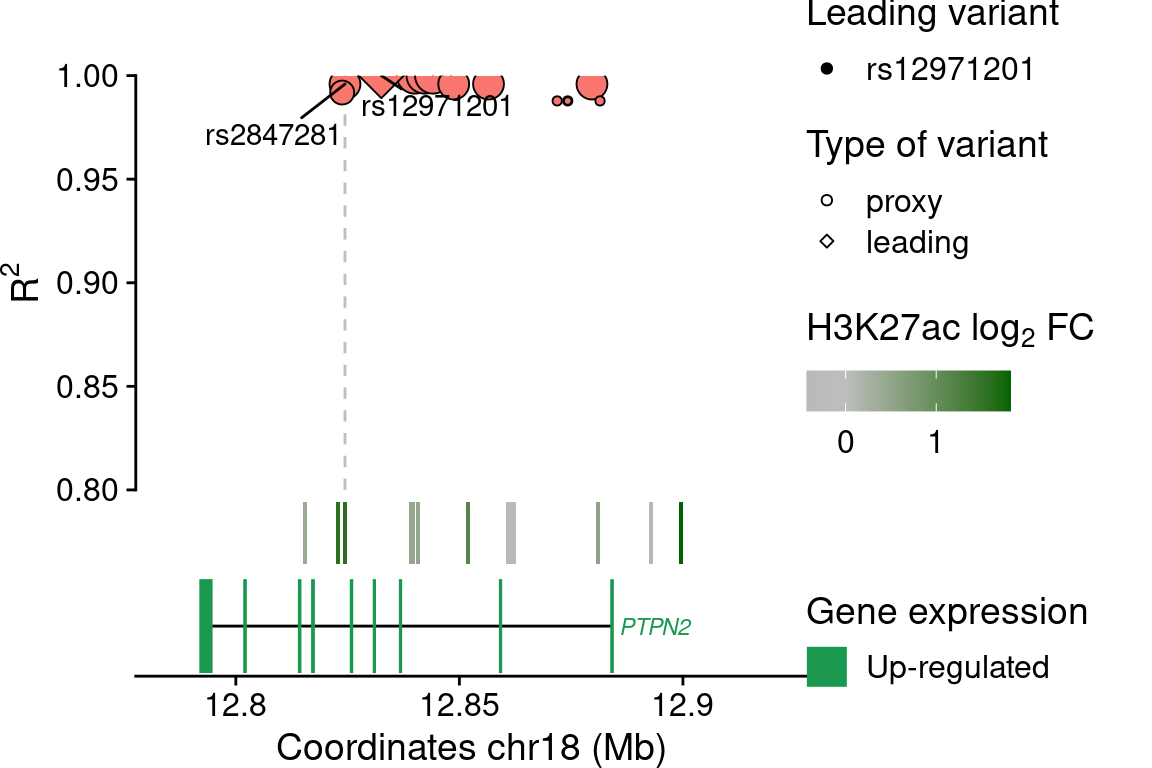
| Version | Author | Date |
|---|---|---|
| 39f446f | mireia-bioinfo | 2020-09-03 |
NUPR1
## SNPs ----------------
rsids <- c("rs4788084")
load("../data/CYT/T1D_GWAS/T1Drm_LD0.8_VSE_AVS.rda")
load("../data/CYT/T1D_GWAS/T1Drm_risk_proxies_granges.rda")
t1d.dat <- data.frame(mcols(proxies))[,c(1,5)]
t1d.dat <- t1d.dat[t1d.dat$R.squared>=0.8,]
snp <- unlist(t1d.avs)
snp <- data.frame(snp[snp$idTag %in% rsids,])
snp$type <- "proxy"
snp$type[snp$idTag==snp$idLd] <- "leading"
snp$lab <- ""
snp$lab[snp$idLd %in% c(rsids, "rs12446550")] <- snp$idLd[snp$idLd %in% c(rsids, "rs12446550")]
snp <- dplyr::left_join(snp,
t1d.dat,
by=c("idLd"="ID"))
snp$type <- factor(snp$type, levels=c("proxy", "leading"))
snp <- snp[order(snp$type),]
win <- GRanges(paste0(unique(snp$seqnames), ":", min(snp$start), "-", max(snp$start)))
win <- resize(win, width=80e3, fix="start")
xlims <- c(start(win), end(win))
## RE --------------------
load("../data/CYT/REs/REs_hi_fc1_padj0.05_granges_subgroup.rda")
re.sel <- subsetByOverlaps(re, win)
re.sel <- resize(re.sel, width=1e3, fix="center")
re.sel <- data.frame(re.sel)
## Genes -------------------
load("../data/CYT/RNA/diffAnalysis/RNA_hi_GRangesBatch.rda")
res.gr$type[res.gr$baseMean<=1] <- "not-expressed"
res.gr <- res.gr[res.gr$gene_biotype=="protein_coding",]
col.df <- data.frame("type"=c(names(pals$differential), "not-expressed"),
"color"=c(pals$differential, "black"),
stringsAsFactors = FALSE)
col.df$color[grep("grey", col.df$color)] <- "grey39"
mcols(res.gr) <- dplyr::left_join(data.frame(mcols(res.gr)[,c(1:2,10)]),
col.df)
colnames(mcols(res.gr))[1] <- "ensembl_gene_id"
mcol.color=4
mcol.name=2
mcol.ensembl=1## Plot snps ------------------
snp.plot <-
ggplot(snp,
aes(start, R.squared)) +
geom_vline(xintercept=snp$start[snp$type=="proxy" & snp$lab!=""],
lty=2, color="grey") +
geom_point(aes(shape=type, size=R.squared, fill=idTag)) +
scale_shape_manual(values=c(21, 23), name="Type of variant") +
scale_size_continuous(guide=F) +
scale_fill_discrete(name="Leading variant") +
ggrepel::geom_text_repel(aes(label=lab)) +
scale_y_continuous(name=expression(R^2),
limits=c(0.8,1),
expand=c(0,0)) +
xlim(xlims) +
themeXblank()
## Plot REs ---------------------
re.plot <-
ggplot(re.sel) +
geom_rect(aes(xmin=start, xmax=end,
ymin=0, ymax=1, fill=h3k27ac.log2FoldChange)) +
scale_fill_gradient2(low="dark grey",
mid="grey",
high="dark green",
name=expression("H3K27ac "*log[2]*" FC"),
breaks=scales::pretty_breaks(n=3),
midpoint=0,
guide = guide_colorbar(direction = "horizontal",
title.position="top")) +
xlim(xlims) +
themeYblank() +
themeXblank()
## Plot genes ------------------
plot.genes <- plotGenes(genes=res.gr[res.gr$type!="not-expressed",],
which=win,
mcol.color=mcol.color,
mcol.name=mcol.name,
mcol.ensembl=mcol.ensembl)
g.plots <-
ggplot() +
plot.genes +
scale_x_continuous(name=paste("Coordinates", seqnames(win), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlims,
labels=function(x) round(x/1e6, 2),
expand=c(0,0)) +
scale_fill_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated")) +
scale_color_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated")) +
themeYblank()
## Create legend --------------
snps.leg <- get_legend(snp.plot)
re.leg <- get_legend(re.plot)
gene.leg <- get_legend(g.plots)
legends <- plot_grid(snps.leg, re.leg, gene.leg, ncol=1)
## Plot all -------------------
main <-
plot_grid(snp.plot + theme(legend.position="none",
plot.margin=margin(1,0,0,0, "cm")),
re.plot + theme(legend.position="none"),
g.plots + theme(legend.position="none"),
ncol=1, align="v", rel_heights=c(0.65, 0.1, 0.25))
plot_grid(main, legends, rel_widths=c(0.7, 0.3))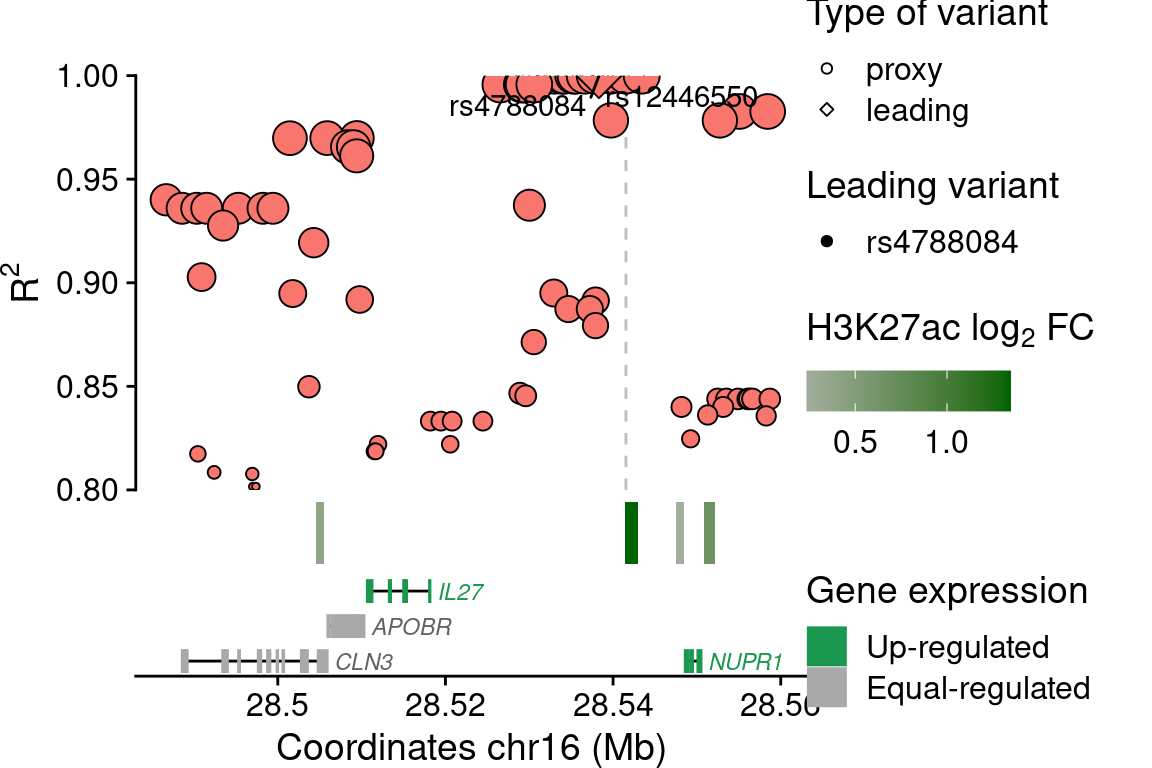
| Version | Author | Date |
|---|---|---|
| 39f446f | mireia-bioinfo | 2020-09-03 |
GSDMB
## SNPs ----------------
rsids <- c("rs12453507")
load("../data/CYT/T1D_GWAS/T1Drm_LD0.8_VSE_AVS.rda")
load("../data/CYT/T1D_GWAS/T1Drm_risk_proxies_granges.rda")
t1d.dat <- data.frame(mcols(proxies))[proxies$topSNP %in% rsids,c(1,5)]
t1d.dat <- t1d.dat[t1d.dat$R.squared>=0.8,]
snp <- unlist(t1d.avs)
snp <- data.frame(snp[snp$idTag %in% rsids,])
row.gr <- proxies[proxies$ID %in% rsids & proxies$CHOSEN,]
row <- data.frame(seqnames=as.character(seqnames(row.gr)),
start=start(row.gr),
end=end(row.gr),
width=1,
strand="*",
idLd=row.gr$ID,
idTag=row.gr$ID)
snp <- rbind(snp, row)
snp$type <- "proxy"
snp$type[snp$idTag==snp$idLd] <- "leading"
snp$lab <- ""
snp$lab[snp$idLd %in% c(rsids, "rs7219923", "rs201389301")] <- snp$idLd[snp$idLd %in% c(rsids, "rs7219923", "rs201389301")]
snp <- dplyr::left_join(snp,
t1d.dat,
by=c("idLd"="ID"))
snp$type <- factor(snp$type, levels=c("proxy", "leading"))
snp <- snp[order(snp$type),]
win <- GRanges(paste0(unique(snp$seqnames), ":", min(snp$start), "-", max(snp$start)))
win <- resize(win, width=30e3, fix="end")
xlims <- c(start(win), end(win))
## RE --------------------
load("../data/CYT/REs/REs_hi_fc1_padj0.05_granges_subgroup.rda")
re.sel <- subsetByOverlaps(re, win)
re.sel <- resize(re.sel, width=1e3, fix="center")
re.sel <- data.frame(re.sel)
## Genes -------------------
load("../data/CYT/RNA/diffAnalysis/RNA_hi_GRangesBatch.rda")
res.gr$type[res.gr$baseMean<=1] <- "not-expressed"
res.gr <- res.gr[res.gr$gene_biotype=="protein_coding",]
col.df <- data.frame("type"=c(names(pals$differential), "not-expressed"),
"color"=c(pals$differential, "black"),
stringsAsFactors = FALSE)
col.df$color[grep("grey", col.df$color)] <- "grey39"
mcols(res.gr) <- dplyr::left_join(data.frame(mcols(res.gr)[,c(1:2,10)]),
col.df)
colnames(mcols(res.gr))[1] <- "ensembl_gene_id"
mcol.color=4
mcol.name=2
mcol.ensembl=1## Plot snps ------------------
snp.plot <-
ggplot(snp,
aes(start, R.squared)) +
geom_vline(xintercept=snp$start[snp$type=="proxy" & snp$lab!=""],
lty=2, color="grey") +
geom_point(aes(shape=type, size=R.squared, fill=idTag)) +
scale_shape_manual(values=c(21, 23), name="Type of variant") +
scale_size_continuous(guide=F) +
scale_fill_discrete(name="Leading variant") +
ggrepel::geom_text_repel(aes(label=lab)) +
scale_y_continuous(name=expression(R^2),
limits=c(0.8,1),
expand=c(0,0)) +
xlim(xlims) +
themeXblank()
## Plot REs ---------------------
re.plot <-
ggplot(re.sel) +
geom_rect(aes(xmin=start, xmax=end,
ymin=0, ymax=1, fill=h3k27ac.log2FoldChange)) +
scale_fill_gradient2(low="dark grey",
mid="grey",
high="dark green",
name=expression("H3K27ac "*log[2]*" FC"),
breaks=scales::pretty_breaks(n=3),
midpoint=0,
limits=c(0,2.5),
guide = guide_colorbar(direction = "horizontal",
title.position="top")) +
xlim(xlims) +
themeYblank() +
themeXblank()
## Plot genes ------------------
plot.genes <- plotGenes(genes=res.gr[res.gr$type!="not-expressed",],
which=win,
mcol.color=mcol.color,
mcol.name=mcol.name,
mcol.ensembl=mcol.ensembl)
g.plots <-
ggplot() +
plot.genes +
scale_x_continuous(name=paste("Coordinates", seqnames(win), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlims,
labels=function(x) round(x/1e6, 2),
expand=c(0,0)) +
scale_fill_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated")) +
scale_color_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated")) +
themeYblank()
## Create legend --------------
snps.leg <- get_legend(snp.plot)
re.leg <- get_legend(re.plot)
gene.leg <- get_legend(g.plots)
legends <- plot_grid(snps.leg, re.leg, gene.leg, ncol=1)
## Plot all -------------------
main <-
plot_grid(snp.plot + theme(legend.position="none",
plot.margin=margin(1,0,0,0, "cm")),
re.plot + theme(legend.position="none"),
g.plots + theme(legend.position="none"),
ncol=1, align="v", rel_heights=c(0.65, 0.1, 0.25))
plot_grid(main, legends, rel_widths=c(0.7, 0.3), scale=c(1,0.5))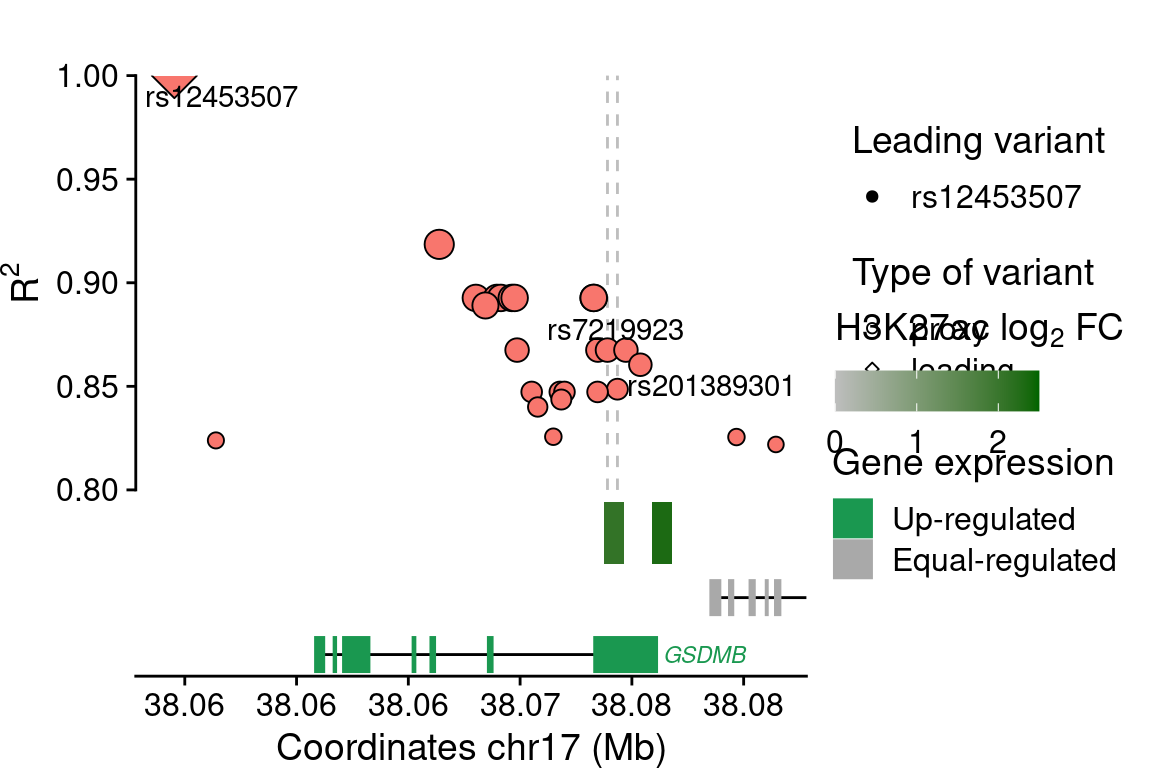
Validated SNP views & Credible Sets
Rscript CYT_getCredibleSets.Rdf <- read.delim("../data/CYT/UMI4C/UMI4C_promoters_views.tsv", stringsAsFactors=T, header=T)
## RE ---------------------------------------------------------------
load("../data/CYT/REs/REs_hi_fc1_padj0.05_granges_subgroup.rda")
## Genes ------------------------------------------------------------
load("../data/CYT/RNA/diffAnalysis/RNA_hi_GRangesBatch.rda")
res.gr$type[res.gr$baseMean<=1] <- "Not-expressed"
res.gr <- res.gr[res.gr$gene_biotype=="protein_coding",]
col.df <- data.frame("type"=c(names(pals$differential), "Not-expressed"),
"color"=c(pals$differential, "black"),
stringsAsFactors = FALSE)
pals$differential <- c(pals$differential, "Not-expressed"="black")
pals["stable"] <- "grey39"
col.df$color[grep("grey", col.df$color)] <- "grey39"
mcols(res.gr) <- unique(dplyr::left_join(data.frame(mcols(res.gr)[,c(1:2,10)]),
col.df,
by=c("type"="type")))
colnames(mcols(res.gr))[1] <- "ensembl_gene_id"
mcol.color=4
mcol.name=2
mcol.ensembl=1rs78037977
i <- grep("rs78037977", df$bait)
load("../output/cyt_04_contacts/UMI4C_norm_results_rs78037977.rda")
xlim <- c(df$start[i], df$end[i])
region <- GRanges(seqnames=paste0(seqnames(res$bait)),
ranges=IRanges(start=xlim[1],
end=xlim[2]))
## CTCF --------------------------------------------------------------
sm <- 200
ctcf <- rtracklayer::import("~/data/TFsIslet_hg19/visualization/CTCF_HI32_hg19-extended_compressed_RPKM.bw",
which=region)
score(ctcf) <- zoo::rollmean(score(ctcf), sm,
fill=c(NA, NA, NA))
plot.ctcf <-
ggplot(data.frame(ctcf)) +
geom_area(aes(x=start,
y=score),
fill="purple3") +
scale_y_continuous(name="CTCF",
limits=c(0,20),
expand=c(0,0),
breaks=c(0,20)) +
scale_x_continuous(name=paste("Coordinates", seqnames(region), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlim,
labels=function(x) round(x/1e6, 2),
expand=c(0,0)) +
themeXblank() +
theme(plot.margin=margin(0.2,0,0.2,0, "cm"))
### Genes ------------------
plot.genes <- plotGenes(genes=res.gr[res.gr$type!="not-expressed",],
which=region,
mcol.color=mcol.color,
mcol.name=mcol.name,
mcol.ensembl=mcol.ensembl)
g.plots <-
ggplot() +
plot.genes +
scale_fill_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated",
`Not-expressed`="Not-expressed")) +
scale_color_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated",
`Not-expressed`="Not-expressed")) +
themeYblank() +
scale_x_continuous(name=paste("Coordinates", seqnames(region), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlim,
labels=function(x) round(x/1e6, 2),
expand=c(0,0))
### UMI-4C ------------------
umi <-
ggplot(res$norm_trend,
aes(start, trend)) +
geom_ribbon(aes(ymin=devM, ymax=devP, group=interaction(group, sample)),
color=NA, fill="light grey") +
geom_line(aes(color=sample, group=interaction(group, sample)),
lwd=0.7) +
annotate("point", x=start(res$bait), y=df$ymax[i], pch=25, fill="black",
size=3) +
annotate("text", x=start(res$bait), y=df$ymax[i]-0.2, label=df$bait[i],
size=3) +
scale_color_manual(values=c(ctrl="#1f78b4", treat="#d95f02"),
labels=c("CYT", "CTRL"), name="Treatment") +
scale_y_continuous(name="# UMI-4C contacts",
limits=c(0, df$ymax[i]),
breaks=scales::pretty_breaks(),
expand=c(0,0)) +
scale_x_continuous(name=paste("Coordinates", seqnames(region), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlim,
labels=function(x) round(x/1e6, 2),
expand=c(0,0)) +
theme(legend.position="right")
### RE ------------------
re.sel <- as.data.frame(subsetByOverlaps(resize(re, 3e3, fix="center"),
region))
ire <-
ggplot(re.sel) +
geom_rect(aes(xmin=start, xmax=end, ymin=0, ymax=1, fill=h3k27ac.log2FoldChange)) +
scale_fill_gradient2(low="dark grey",
mid="grey",
high="dark green",
name=expression("H3K27ac "*log[2]*" FC"),
breaks=scales::pretty_breaks(n=3),
midpoint=0,
guide = guide_colorbar(direction = "horizontal",
title.position="top")) +
scale_x_continuous(name=paste("Coordinates", seqnames(region), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlim,
labels=function(x) round(x/1e6, 2),
expand=c(0,0))
### Get legends -----------
gene.leg <- get_legend(g.plots)
umi.leg <- get_legend(umi)
ire.leg <- get_legend(ire)
legends <- plot_grid(umi.leg, ire.leg, gene.leg, ncol=1)
### Grid ------------------
p <-
plot_grid(umi + themeXblank() + theme(legend.position = "none",
plot.margin=margin(1,0,0,0, "cm")),
ire + themeYblank() + themeXblank() + theme(legend.position = "none"),
plot.ctcf,
g.plots + theme(legend.position = "none"),
ncol=1,
rel_heights = c(0.5, 0.08, 0.12, 0.3),
align="v")
plot <-
plot_grid(p,
legends,
ncol=2, rel_widths=c(0.7, 0.3))
plot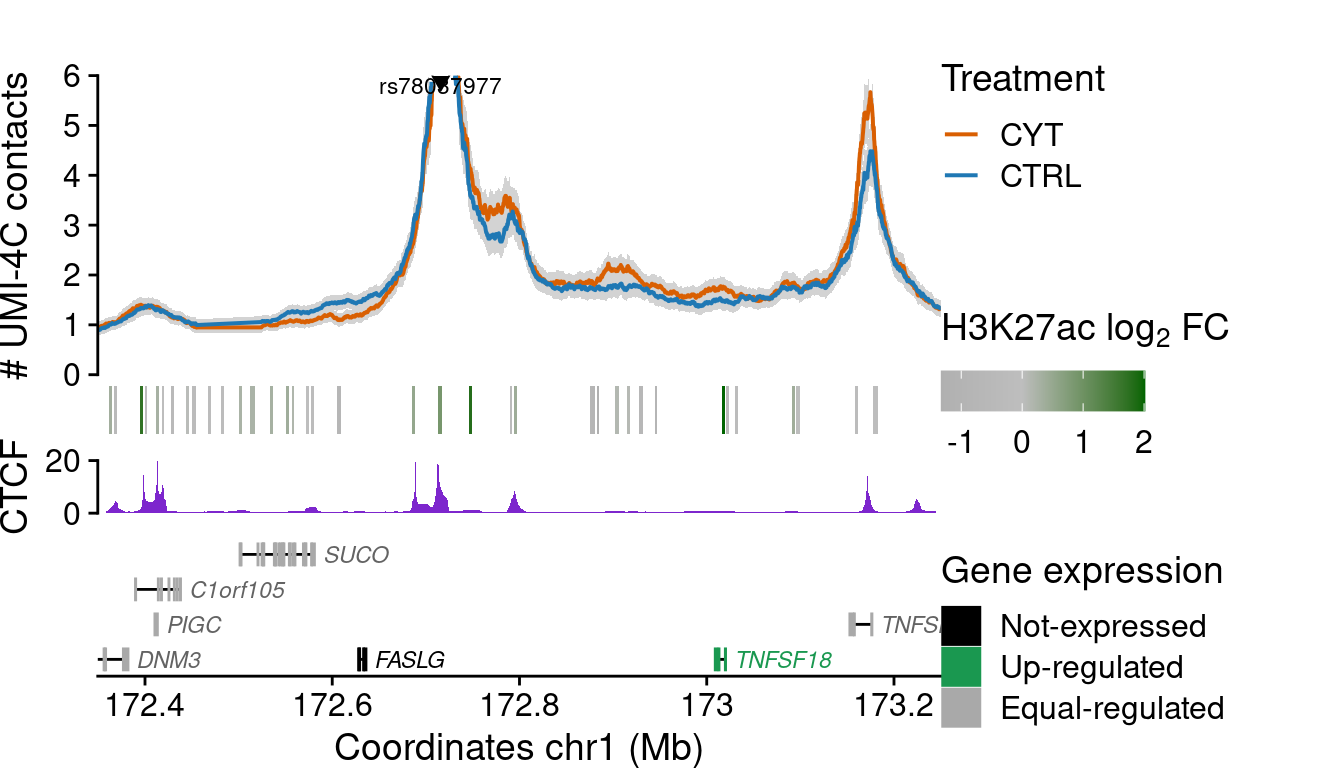
Zoom ins
win <- 7e3
snp <- GRanges("chr1:172715702")
snp <- resize(snp, width=win, fix="center")
##-------------------------------
## Load tracks
##-------------------------------
## ATAC-seq
sm.at <- 5
ctrl.at <- rtracklayer::import("../data/CYT/ATAC/Visualization/ATAC_hi_ctrl.bw",
which=snp)
score(ctrl.at) <- zoo::rollmean(score(ctrl.at), sm.at,
fill=c(NA, NA, NA))
cyt.at <- rtracklayer::import("../data/CYT/ATAC/Visualization/ATAC_hi_cyt.bw",
which=snp)
score(cyt.at) <- zoo::rollmean(score(cyt.at), sm.at,
fill=c(NA, NA, NA))
## H3K27ac
sm.ac <- 5
ctrl.ac <- rtracklayer::import("../data/CYT/H3K27ac/Visualization/H3K27ac_hi_ctrl.bw",
which=snp)
score(ctrl.ac) <- zoo::rollmean(score(ctrl.ac), sm.ac,
fill=c(NA, NA, NA))
cyt.ac <- rtracklayer::import("../data/CYT/H3K27ac/Visualization/H3K27ac_hi_cyt.bw",
which=snp)
score(cyt.ac) <- zoo::rollmean(score(cyt.ac), sm.ac,
fill=c(NA, NA, NA))
## Islet TFS
files <- list.files("~/data/TFsIslet_hg19/visualization",
pattern=".bw", full.names=TRUE)[-1]
islet <- lapply(files, rtracklayer::import,
which=snp)
for (i in 1:length(islet)) {
score(islet[[i]]) <- zoo::rollmean(score(islet[[i]]), 10,
fill=c(NA, NA, NA))
}
names <- pipelineNGS::getNameFromPath(files, suffix=".bw")
len <- sapply(islet, length)
islet <- GRangesList(islet)
islet <- unlist(islet)
islet$TF <- unlist(mapply(rep, names, each=len))
## RE
load("../data/CYT/REs/REs_hi_fc1_padj0.05_granges_subgroup.rda")
re.sel <- subsetByOverlaps(re, snp)
## Conservation
cons <- rtracklayer::import("~/data/phastCons_46_placentalMammals/placental_mammals.bw",
which=snp)
load("../data/CYT/T1D_credibleSet/uk_credibleSet_fullr2.rda")
uk.full <- GRanges(uk.full)
uk <- subsetByOverlaps(uk.full,snp)
uk$lab <- ""
uk$lab[uk$ID=="rs78037977"] <- uk$ID[uk$ID=="rs78037977"]##-------------------------------
## Plot tracks
##-------------------------------
xlims <- c(start(ranges(snp)),
end(ranges(snp)))
cons.p <-
ggplot(data.frame(cons)) +
geom_area(aes(x=start,
y=score),
fill="maroon4") +
scale_y_continuous(name="",
limits=c(0,1),
expand=c(0,0),
breaks=c(0,1)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
ctrl.at.p <-
ggplot(data.frame(ctrl.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,60),
expand=c(0,0),
breaks=c(0,60)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.at.p <-
ggplot(data.frame(cyt.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,60),
expand=c(0,0),
breaks=c(0,60)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
ctrl.ac.p <-
ggplot(data.frame(ctrl.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,20),
expand=c(0,0),
breaks=c(0,20)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.ac.p <-
ggplot(data.frame(cyt.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,20),
expand=c(0,0),
breaks=c(0,20)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
islet.plot <-
ggplot(data.frame(islet)) +
geom_area(aes(x=start,
y=score,
fill=TF),
alpha=0.6, position="identity") +
scale_y_continuous(name="",
limits=c(0,10),
expand=c(0,0),
breaks=c(0,10)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"),
legend.position="none")
re.plot <-
ggplot(data.frame(re.sel)) +
geom_rect(aes(xmin=start, xmax=end,
ymin=0, ymax=1, fill=h3k27ac.log2FoldChange),
color="black") +
scale_fill_gradient(low=pals$differential["stable"],
high=pals$differential["gained"],
limits=c(0,3)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeYblank() +
themeXblank() +
theme(legend.position="none")
snp.plot <-
ggplot(data.frame(uk)) +
geom_vline(xintercept=172715702, lty=2) +
geom_point(aes(start, -log10(pmeta),
pch=inCredible,
size=r2, fill=r2),
color="black") +
scale_shape_manual(values=c(21,23),
guide=FALSE) +
geom_text(aes(start, -log10(pmeta),
label=lab),
alpha=0.8) +
scale_fill_gradient2(low="white",
high="goldenrod3",
guide=FALSE) +
scale_size_continuous(range=c(0.05, 4),
guide=FALSE) +
scale_y_continuous(name=expression(-Log[10]*" P"),
breaks=scales::pretty_breaks(n=4),
expand=c(0,0),
limits=c(0,NA)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
plot_grid(cons.p,
islet.plot,
ctrl.at.p,
cyt.at.p,
ctrl.ac.p,
cyt.ac.p,
snp.plot,
re.plot,
ncol=1,
rel_heights=c(rep(0.12, 6), 0.24, 0.04),
align="v")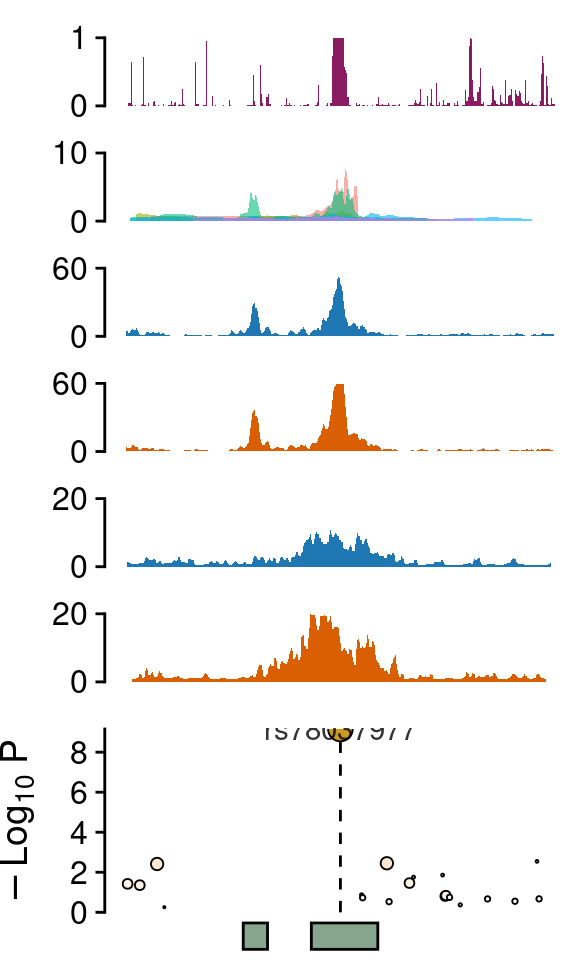
| Version | Author | Date |
|---|---|---|
| 39f446f | mireia-bioinfo | 2020-09-03 |
load("../data/CYT/RNA/diffAnalysis/RNA_hi_GRangesBatch.rda")
gene <- res.gr[res.gr$external_gene_name=="TNFSF18",]
gene <- resize(gene, width=2e4, fix="center")
gene$color <- pals$differential["gained"]
colnames(mcols(gene))[1] <- "ensembl_gene_id"
##-------------------------------
## Load tracks
##-------------------------------
## ATAC-seq
sm.at <- 20
ctrl.at <- rtracklayer::import("../data/CYT/ATAC/Visualization/ATAC_hi_ctrl.bw",
which=gene)
score(ctrl.at) <- zoo::rollmean(score(ctrl.at), sm.at,
fill=c(NA, NA, NA))
cyt.at <- rtracklayer::import("../data/CYT/ATAC/Visualization/ATAC_hi_cyt.bw",
which=gene)
score(cyt.at) <- zoo::rollmean(score(cyt.at), sm.at,
fill=c(NA, NA, NA))
## H3K27ac
sm.ac <- 20
ctrl.ac <- rtracklayer::import("../data/CYT/H3K27ac/Visualization/H3K27ac_hi_ctrl.bw",
which=gene)
score(ctrl.ac) <- zoo::rollmean(score(ctrl.ac), sm.ac,
fill=c(NA, NA, NA))
cyt.ac <- rtracklayer::import("../data/CYT/H3K27ac/Visualization/H3K27ac_hi_cyt.bw",
which=gene)
score(cyt.ac) <- zoo::rollmean(score(cyt.ac), sm.ac,
fill=c(NA, NA, NA))
## RNA-seq
sm.rna <- 20
ctrl.rna <- rtracklayer::import("../data/CYT/RNA/Visualization/RNA_hi_ctrl.bw",
which=gene)
score(ctrl.rna) <- zoo::rollmean(score(ctrl.rna), sm.rna,
fill=c(NA, NA, NA))
cyt.rna <- rtracklayer::import("../data/CYT/RNA/Visualization/RNA_hi_cyt.bw",
which=gene)
score(cyt.rna) <- zoo::rollmean(score(cyt.rna), sm.rna,
fill=c(NA, NA, NA))
## RE
load("../data/CYT/REs/REs_hi_fc1_padj0.05_granges_subgroup.rda")
re.sel <- subsetByOverlaps(re, gene)##-------------------------------
## Plot tracks
##-------------------------------
xlims <- c(start(ranges(gene)),
end(ranges(gene)))
ctrl.at.p <-
ggplot(data.frame(ctrl.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,30),
expand=c(0,0),
breaks=c(0,30)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.at.p <-
ggplot(data.frame(cyt.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,30),
expand=c(0,0),
breaks=c(0,30)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
ctrl.ac.p <-
ggplot(data.frame(ctrl.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,15),
expand=c(0,0),
breaks=c(0,15)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.ac.p <-
ggplot(data.frame(cyt.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,15),
expand=c(0,0),
breaks=c(0,15)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
ctrl.rna.p <-
ggplot(data.frame(ctrl.rna)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,3),
expand=c(0,0),
breaks=c(0,3)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.rna.p <-
ggplot(data.frame(cyt.rna)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,3),
expand=c(0,0),
breaks=c(0,3)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
genes.p <-
ggplot() +
plotGenes(gene,
gene,
mcol.color=11,
mcol.name=2,
mcol.ensembl=1)
genes.p <-
genes.p +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
theme(legend.position="none") +
themeYblank() +
themeXblank()
plot_grid(ctrl.at.p,
cyt.at.p,
ctrl.ac.p,
cyt.ac.p,
ctrl.rna.p,
cyt.rna.p,
genes.p,
ncol=1,
rel_heights=c(rep(0.16, 6), 0.04),
align="v")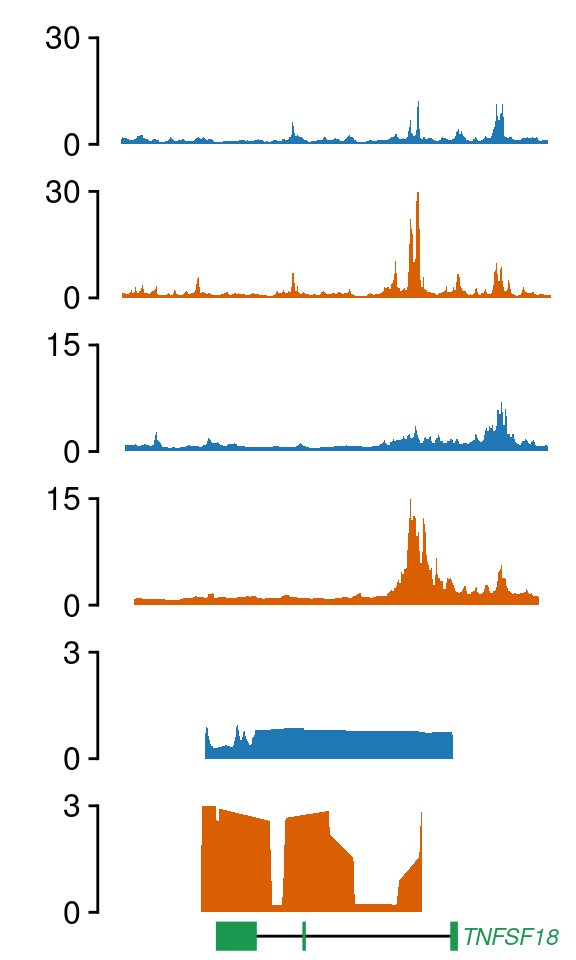
| Version | Author | Date |
|---|---|---|
| 39f446f | mireia-bioinfo | 2020-09-03 |
DEXI
## RE ---------------------------------------------------------------
load("../data/CYT/REs/REs_hi_fc1_padj0.05_granges_subgroup.rda")
## Genes ------------------------------------------------------------
load("../data/CYT/RNA/diffAnalysis/RNA_hi_GRangesBatch.rda")
res.gr$type[res.gr$baseMean<=1] <- "Not-expressed"
res.gr <- res.gr[res.gr$gene_biotype=="protein_coding",]
col.df <- data.frame("type"=c(names(pals$differential), "Not-expressed"),
"color"=c(pals$differential, "black"),
stringsAsFactors = FALSE)
pals$differential <- c(pals$differential, "Not-expressed"="black")
pals["stable"] <- "grey39"
col.df$color[grep("grey", col.df$color)] <- "grey39"
mcols(res.gr) <- unique(dplyr::left_join(data.frame(mcols(res.gr)[,c(1:2,10)]),
col.df))
colnames(mcols(res.gr))[1] <- "ensembl_gene_id"
mcol.color=4
mcol.name=2
mcol.ensembl=1i <- grep("DEXI", df$bait)
load(paste0("../output/cyt_04_contacts/UMI4C_norm_results_DEXI.rda"))
load("../data/CYT/T1D_credibleSet/T1D_credibleSet_fullR2_rs193778.rda")
uk$lab <- ""
uk$lab[grep("rs193778", uk$ID)] <- "rs193778"
uk <- uk[order(uk$inCredible),]
xlim <- c(df$start[i], df$end[i])
region <- GRanges(seqnames=paste0(seqnames(res$bait)),
ranges=IRanges(start=xlim[1],
end=xlim[2]))
## H3K27ac --------------------------------------------------------------
sm <- 100
ctrl <- rtracklayer::import("../data/CYT/H3K27ac/Visualization/H3K27ac_hi_ctrl.bw",
which=region)
score(ctrl) <- zoo::rollmean(score(ctrl), sm,
fill=c(NA, NA, NA))
plot.ctrl <-
ggplot(data.frame(ctrl)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="H3K27ac",
limits=c(0,50),
expand=c(0,0),
breaks=c(0,50)) +
scale_x_continuous(name=paste("Coordinates", seqnames(region), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlim,
labels=function(x) round(x/1e6, 2),
expand=c(0,0)) +
themeXblank() +
theme(plot.margin=margin(0.2,0,0.2,0, "cm"))
cyt <- rtracklayer::import("../data/CYT/H3K27ac/Visualization/H3K27ac_hi_cyt.bw",
which=region)
score(cyt) <- zoo::rollmean(score(cyt), sm,
fill=c(NA, NA, NA))
plot.cyt <-
ggplot(data.frame(cyt)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="H3K27ac",
limits=c(0,50),
expand=c(0,0),
breaks=c(0,50)) +
scale_x_continuous(name=paste("Coordinates", seqnames(region), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlim,
labels=function(x) round(x/1e6, 2),
expand=c(0,0)) +
themeXblank() +
theme(plot.margin=margin(0.2,0,0.2,0, "cm"))
### Genes ------------------
colnames(mcols(res.gr))[1] <- "ensembl_gene_id"
plot.genes <- plotGenes(genes=res.gr[res.gr$type!="not-expressed",],
which=region,
mcol.color=mcol.color,
mcol.name=mcol.name,
mcol.ensembl=mcol.ensembl)
g.plots <-
ggplot() +
plot.genes +
xlim(xlim) +
scale_fill_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated",
`Not-expressed`="Not-expressed")) +
scale_color_manual(values=pals$differential,
name="Gene expression",
labels=c(gained="Up-regulated",
lost="Down-regulated",
stable="Equal-regulated",
`Not-expressed`="Not-expressed")) +
themeYblank() +
scale_x_continuous(name=paste("Coordinates", seqnames(region), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlim,
labels=function(x) round(x/1e6, 2),
expand=c(0,0))
### UMI-4C ------------------
umi <-
ggplot(res$norm_trend,
aes(start, trend)) +
geom_ribbon(aes(ymin=devM, ymax=devP, group=interaction(group, sample)),
color=NA, fill="light grey") +
geom_line(aes(color=sample, group=interaction(group, sample)),
lwd=0.7) +
annotate("point", x=start(res$bait), y=df$ymax[i], pch=25, fill="black",
size=3) +
annotate("text", x=start(res$bait), y=df$ymax[i]-0.2, label=df$bait[i],
size=3) +
geom_point(data=uk,
aes(start, scales::rescale(-log10(pmeta), to=c(0, df$ymax[i])),
fill=r2, shape=inCredible,
alpha=scales::rescale(-log10(pmeta), to=c(0, df$ymax[i])),
size=r2)) +
ggrepel::geom_text_repel(data=uk,
aes(start, scales::rescale(-log10(pmeta), to=c(0, df$ymax[i])),
label=lab)) +
geom_vline(xintercept=uk$start[uk$lab!=""], lty=2, color="grey") +
scale_color_manual(values=c(ctrl="#1f78b4", treat="#d95f02"),
labels=c("CYT", "CTRL"), name="Treatment") +
scale_alpha_continuous(range=c(0.05, 0.7), guide=FALSE) +
scale_fill_gradient2(low="white", high="goldenrod3",
limits=c(0,1), breaks=c(0,1),
name=expression(R^2),
guide=guide_colorbar(direction = "horizontal",
title.position="top")) +
scale_shape_manual(values=c(21,23)) +
scale_size_continuous(range=c(0.1,5)) +
scale_y_continuous(name="# UMI-4C contacts",
limits=c(0, df$ymax[i]),
breaks=scales::pretty_breaks(),
expand=c(0,0),
sec.axis = sec_axis(~.*(max(-log10(uk$pmeta)/df$ymax[i])),
name=expression(-Log[10]*" P"))) +
scale_x_continuous(name=paste("Coordinates", seqnames(region), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlim,
labels=function(x) round(x/1e6, 2),
expand=c(0,0)) +
theme(legend.position="right") +
guides(alpha=F, size=F, shape=F)
### RE ------------------
re.sel <- as.data.frame(subsetByOverlaps(resize(re, 3e3, fix="center"),
region))
ire <-
ggplot(re.sel) +
geom_rect(aes(xmin=start, xmax=end, ymin=0, ymax=1, fill=h3k27ac.log2FoldChange)) +
scale_fill_gradient2(low="dark grey",
mid="grey",
high="dark green",
name=expression("H3K27ac "*log[2]*" FC"),
breaks=scales::pretty_breaks(n=3),
midpoint=0,
guide = guide_colorbar(direction = "horizontal",
title.position="top")) +
scale_x_continuous(name=paste("Coordinates", seqnames(region), "(Mb)"),
breaks=scales::pretty_breaks(),
limits=xlim,
labels=function(x) round(x/1e6, 2),
expand=c(0,0))
### Get legends -----------
gene.leg <- get_legend(g.plots)
umi.leg <- get_legend(umi)
ire.leg <- get_legend(ire)
legends <- plot_grid(umi.leg, ire.leg, gene.leg, ncol=1)
### Grid ------------------
p <-
plot_grid(umi + themeXblank() + theme(legend.position = "none",
plot.margin=margin(1,0,0,0, "cm")),
ire + themeYblank() + themeXblank() + theme(legend.position = "none"),
plot.ctrl,
plot.cyt,
g.plots + theme(legend.position = "none"),
ncol=1,
rel_heights = c(0.45, 0.05, 0.08, 0.08, 0.25),
align="v")
plot <-
plot_grid(p,
legends,
ncol=2, rel_widths=c(0.7, 0.3))
plot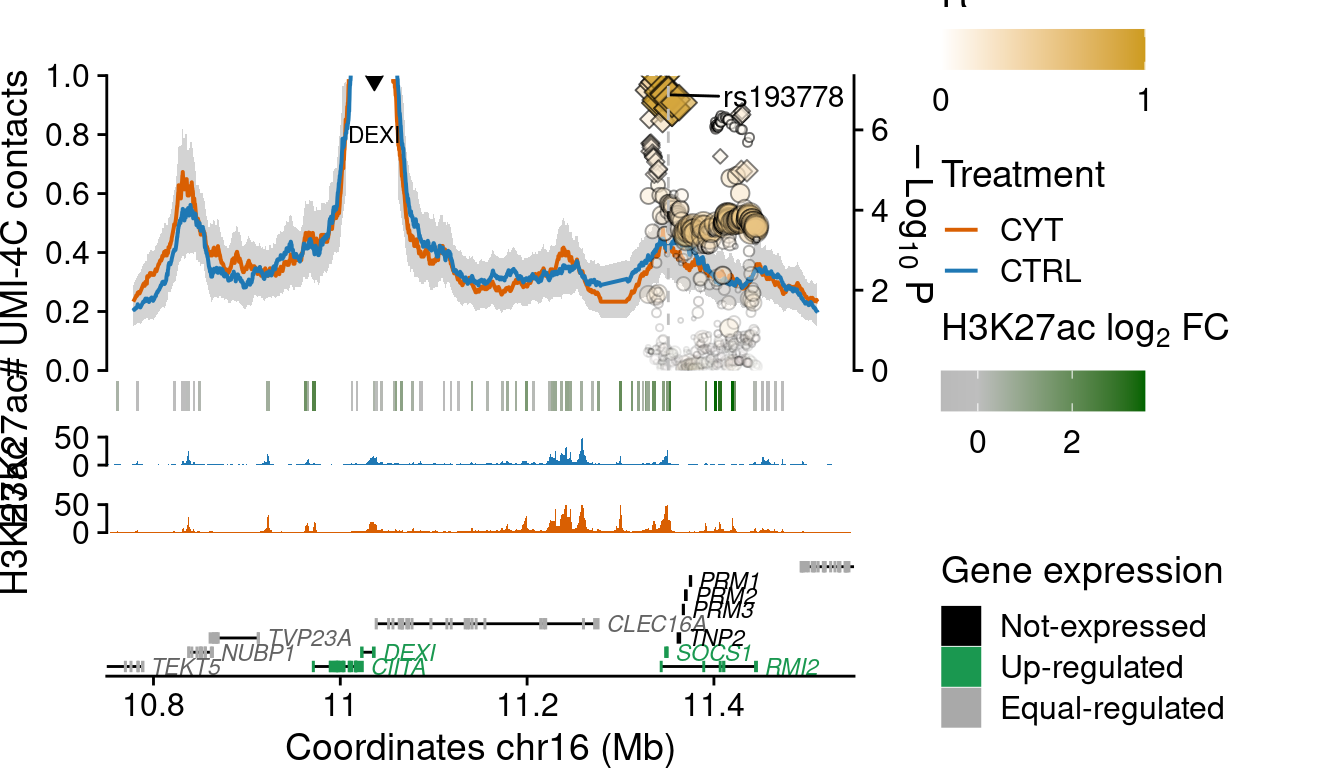
| Version | Author | Date |
|---|---|---|
| 39f446f | mireia-bioinfo | 2020-09-03 |
Zoom ins
win <- 8e2
snp <- GRanges("chr16:11351211")
snp <- resize(snp, width=win, fix="center")
##-------------------------------
## Load tracks
##-------------------------------
## ATAC-seq
sm.at <- 5
ctrl.at <- rtracklayer::import("../data/CYT/ATAC/Visualization/ATAC_hi_ctrl.bw",
which=snp)
score(ctrl.at) <- zoo::rollmean(score(ctrl.at), sm.at,
fill=c(NA, NA, NA))
cyt.at <- rtracklayer::import("../data/CYT/ATAC/Visualization/ATAC_hi_cyt.bw",
which=snp)
score(cyt.at) <- zoo::rollmean(score(cyt.at), sm.at,
fill=c(NA, NA, NA))
## H3K27ac
sm.ac <- 5
ctrl.ac <- rtracklayer::import("../data/CYT/H3K27ac/Visualization/H3K27ac_hi_ctrl.bw",
which=snp)
score(ctrl.ac) <- zoo::rollmean(score(ctrl.ac), sm.ac,
fill=c(NA, NA, NA))
cyt.ac <- rtracklayer::import("../data/CYT/H3K27ac/Visualization/H3K27ac_hi_cyt.bw",
which=snp)
score(cyt.ac) <- zoo::rollmean(score(cyt.ac), sm.ac,
fill=c(NA, NA, NA))
## Islet TFS
files <- list.files("~/data/TFsIslet_hg19/visualization",
pattern=".bw", full.names=TRUE)[-1]
islet <- lapply(files, rtracklayer::import,
which=snp)
for (i in 1:length(islet)) {
score(islet[[i]]) <- zoo::rollmean(score(islet[[i]]), 10,
fill=c(NA, NA, NA))
}
names <- pipelineNGS::getNameFromPath(files, suffix=".bw")
len <- sapply(islet, length)
islet <- GRangesList(islet)
islet <- unlist(islet)
islet$TF <- unlist(mapply(rep, names, each=len))
## RE
load("../data/CYT/REs/REs_hi_fc1_padj0.05_granges_subgroup.rda")
re.sel <- subsetByOverlaps(re, snp)
## Conservation
cons <- rtracklayer::import("~/data/phastCons_46_placentalMammals/placental_mammals.bw",
which=snp)
load("../data/CYT/T1D_credibleSet/uk_credibleSet_fullr2.rda")
uk.full <- GRanges(uk.full)
uk <- subsetByOverlaps(uk.full,snp)
uk$lab <- ""
uk$lab[uk$ID=="rs193778"] <- uk$ID[uk$ID=="rs193778"]##-------------------------------
## Plot tracks
##-------------------------------
xlims <- c(start(ranges(snp)),
end(ranges(snp)))
cons.p <-
ggplot(data.frame(cons)) +
geom_area(aes(x=start,
y=score),
fill="maroon4") +
scale_y_continuous(name="",
limits=c(0,1),
expand=c(0,0),
breaks=c(0,1)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
ctrl.at.p <-
ggplot(data.frame(ctrl.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,50),
expand=c(0,0),
breaks=c(0,50)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.at.p <-
ggplot(data.frame(cyt.at)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,50),
expand=c(0,0),
breaks=c(0,50)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
ctrl.ac.p <-
ggplot(data.frame(ctrl.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["ctrl"]) +
scale_y_continuous(name="",
limits=c(0,50),
expand=c(0,0),
breaks=c(0,50)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
cyt.ac.p <-
ggplot(data.frame(cyt.ac)) +
geom_area(aes(x=start,
y=score),
fill=pals$treatment["cyt"]) +
scale_y_continuous(name="",
limits=c(0,50),
expand=c(0,0),
breaks=c(0,50)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
islet.plot <-
ggplot(data.frame(islet)) +
geom_area(aes(x=start,
y=score,
fill=TF),
alpha=0.6, position="identity") +
scale_y_continuous(name="",
limits=c(0,5),
expand=c(0,0),
breaks=c(0,5)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"),
legend.position="none")
re.plot <-
ggplot(data.frame(re.sel)) +
geom_rect(aes(xmin=start, xmax=end,
ymin=0, ymax=1, fill=h3k27ac.log2FoldChange),
color="black") +
scale_fill_gradient(low=pals$differential["stable"],
high=pals$differential["gained"],
limits=c(0,3)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeYblank() +
themeXblank() +
theme(legend.position="none")
snp.plot <-
ggplot(data.frame(uk)) +
geom_vline(xintercept=start(uk[uk$lab!=""]), lty=2) +
geom_point(aes(start, -log10(pmeta),
pch=inCredible,
size=r2, fill=r2),
color="black") +
scale_shape_manual(values=c(21,23),
guide=FALSE) +
geom_text(aes(start, -log10(pmeta),
label=lab),
alpha=0.8) +
scale_fill_gradient2(low="white",
high="goldenrod3",
guide=FALSE) +
scale_size_continuous(range=c(0.05, 4),
guide=FALSE) +
scale_y_continuous(name=expression(-Log[10]*" P"),
breaks=scales::pretty_breaks(n=4),
expand=c(0,0),
limits=c(0,NA)) +
scale_x_continuous(limits=xlims,
labels=function(x) round(x/1e6, 3),
breaks=scales::pretty_breaks(n=2),
name="Coordinates (Mb)") +
themeXblank() +
theme(plot.margin=unit(c(0.5,0,0,0), "cm"))
plot_grid(cons.p,
islet.plot,
ctrl.at.p,
cyt.at.p,
ctrl.ac.p,
cyt.ac.p,
snp.plot,
re.plot,
ncol=1,
rel_heights=c(rep(0.12, 6), 0.24, 0.04),
align="v")
sessionInfo()R version 4.0.2 (2020-06-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.1 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=es_ES.UTF-8 LC_COLLATE=C
[5] LC_MONETARY=es_ES.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=es_ES.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=es_ES.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] umi4cCatcheR_0.0.0.9000 VSE_0.99 dplyr_1.0.1
[4] kableExtra_1.1.0 cowplot_1.0.0 ggplot2_3.3.2
[7] GenomicRanges_1.41.5 GenomeInfoDb_1.25.8 IRanges_2.23.10
[10] S4Vectors_0.27.12 BiocGenerics_0.35.4 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] colorspace_1.4-1 ellipsis_0.3.1
[3] rio_0.5.16 rprojroot_1.3-2
[5] XVector_0.29.3 fs_1.5.0
[7] rstudioapi_0.11 farver_2.0.3
[9] remotes_2.2.0 bit64_4.0.2
[11] ggrepel_0.8.2 AnnotationDbi_1.51.3
[13] fansi_0.4.1 xml2_1.3.2
[15] knitr_1.29 pkgload_1.1.0
[17] Rsamtools_2.5.3 dbplyr_1.4.4
[19] pipelineNGS_0.0.0.9000 readr_1.3.1
[21] compiler_4.0.2 httr_1.4.2
[23] backports_1.1.8 Matrix_1.2-18
[25] assertthat_0.2.1 cli_2.0.2
[27] later_1.1.0.1 htmltools_0.5.0
[29] prettyunits_1.1.1 tools_4.0.2
[31] igraph_1.2.5 gtable_0.3.0
[33] glue_1.4.1 GenomeInfoDbData_1.2.3
[35] rappdirs_0.3.1 Rcpp_1.0.5
[37] Biobase_2.49.0 carData_3.0-4
[39] cellranger_1.1.0 vctrs_0.3.2
[41] Biostrings_2.57.2 rtracklayer_1.49.4
[43] xfun_0.16 stringr_1.4.0
[45] ps_1.3.3 openxlsx_4.1.5
[47] testthat_2.3.2 rvest_0.3.6
[49] lifecycle_0.2.0 devtools_2.3.1
[51] XML_3.99-0.5 zoo_1.8-8
[53] zlibbioc_1.35.0 scales_1.1.1
[55] BSgenome_1.57.5 hms_0.5.3
[57] promises_1.1.1 SummarizedExperiment_1.19.6
[59] yaml_2.2.1 curl_4.3
[61] memoise_1.1.0 gridExtra_2.3
[63] biomaRt_2.45.2 RSQLite_2.2.0
[65] stringi_1.4.6 highr_0.8
[67] desc_1.2.0 pkgbuild_1.1.0
[69] zip_2.0.4 BiocParallel_1.23.2
[71] rlang_0.4.7 pkgconfig_2.0.3
[73] bitops_1.0-6 matrixStats_0.56.0
[75] lattice_0.20-41 evaluate_0.14
[77] purrr_0.3.4 GenomicAlignments_1.25.3
[79] labeling_0.3 bit_4.0.4
[81] processx_3.4.3 tidyselect_1.1.0
[83] magrittr_1.5 bookdown_0.20
[85] R6_2.4.1 generics_0.0.2
[87] DBI_1.1.0 DelayedArray_0.15.7
[89] pillar_1.4.6 haven_2.3.1
[91] whisker_0.4 foreign_0.8-80
[93] withr_2.2.0 abind_1.4-5
[95] RCurl_1.98-1.2 tibble_3.0.3
[97] crayon_1.3.4 car_3.0-8
[99] BiocFileCache_1.13.1 rmarkdown_2.3
[101] progress_1.2.2 usethis_1.6.1
[103] grid_4.0.2 readxl_1.3.1
[105] data.table_1.13.0 blob_1.2.1
[107] callr_3.4.3 git2r_0.27.1
[109] forcats_0.5.0 digest_0.6.25
[111] webshot_0.5.2 httpuv_1.5.4
[113] regioneR_1.21.1 openssl_1.4.2
[115] munsell_0.5.0 viridisLite_0.3.0
[117] askpass_1.1 sessioninfo_1.1.1